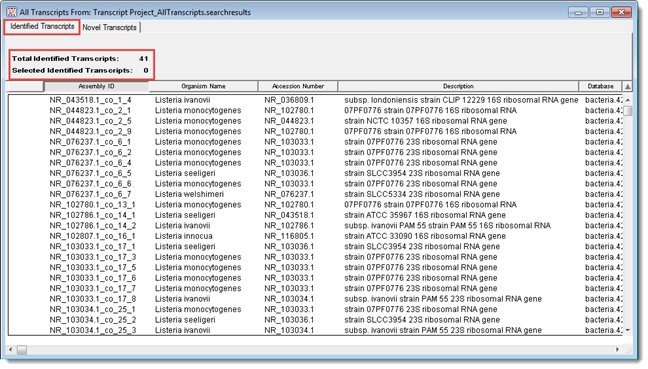
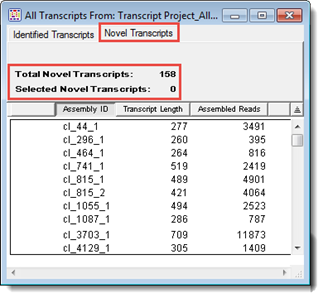
Once you have created a de novo transcriptome assembly in SeqMan NGen and opened it in SeqMan Pro, the All Transcripts window is shown. This window contains two tabs: one displays the Identified Transcripts table, and the other the Novel Transcripts table. The heading for each tab shows the total number of transcripts in the table, and the number currently selected.
The Identified Transcripts table shows transcripts that had a Transcript Annotation Database match exceeding the specified thresholds. These are labeled with information from the best matching database entry following the default convention: [gene name]_[accession]_co_[assembly ID]_[contig ID].
The Novel Transcripts table shows assembled transcripts that either did not have a database match or had a preliminary match that then fell below thresholds upon further processing. The former is labeled following the convention cl_[assembly ID]_[contig ID] and the latter as [gene name]_[accession]_co_[assembly ID]_[contig ID] to give a hint as to the possible identity of that sequence.
The following tasks can be done within the Transcript Tables:
|
Task |
How to… |
|
Display, hide, rearrange or sort data columns |
See Sorting and Rearranging Tables. That help topic also discusses the meaning of the “rainbow” coloring that occurs when you sort the Identified Transcripts table. For a list of available data columns, see Available Transcript Table Columns. |
|
Open individual contigs for visualization and editing |
Double click on the row of interest, and the appropriate .sqd document will be loaded with the Alignment View of the selected contig displayed. All the usual visualization and editing tools in SeqMan Pro can be used. Note that edits will not be reflected in the AllTranscripts.searchresults table, nor will the consensus sequence for the edited contig(s) be updated in the multi-sequence .fasta file in the .Transcripts package.
Transcripts that have been deleted, or that have been merged into other transcripts using the Align > Align Contigs command, will remain in the table and the multi-sequence .fasta file. Double-clicking on any of the entries will elicit a warning that the transcript no longer exists. If you save the corresponding .sqd document following editing, any changes will be saved. |
|
Save a subset of transcripts for downstream use with SeqMan NGen |
|
|
Use a context menu to access editing commands |