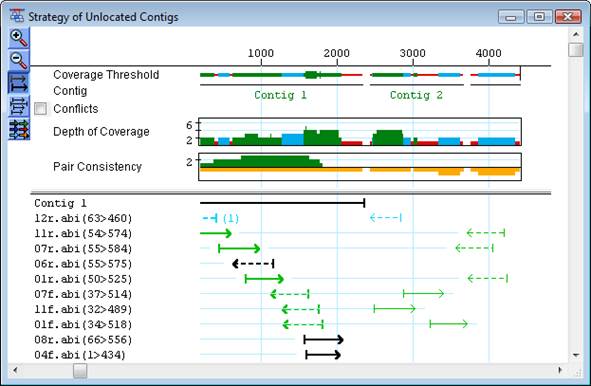
Paired end sequence data are forward and reverse sequence reads originating at opposite ends of the same fragment. Even though such a pair of reads may not overlap by sequence similarity, they belong in the same contig. If the reads are assembled into different contigs, the knowledge they are linked may help in joining two contigs together though it may instead indicate that the reads are misassembled or misnamed. Paired end data can also be used for within-contig assembly evaluation. If the two reads for a given pair are not in opposite orientations, for instance, an assembly problem may be indicated. Finally, if the distance between the two reads is known (+/-), this information can also be used to confirm whether or not the assembly is correct. When using paired end sequence data to evaluate assemblies, be sure to allow for the possibility that there may be errors in some of the sequence names.
Before you can use paired end data, SeqMan Pro needs to recognize your sequence naming convention, as well as your specifications for a minimum and maximum distance between the opposite ends of your inserts. Use the Pair Specifier parameters to define these values.
Ideally, paired end sequencing projects should use a simple, consistent, perfectly-implemented naming convention for forward and reverse reads. However, this goal may not be achieved in every project. The pair specifier in SeqMan Pro has enormous flexibility for creating expressions that will still distinguish paired end data when naming conventions are complex or imperfect.
Note: Since paired end information is used only after assembly, bad information about paired reads cannot poison the assembly. This is critical for data obtained by gel sequencing, as lane tracking errors sometimes lead to misnaming of a proportion of reads in a larger project. Therefore, paired read information that the software displays as inconsistent needs careful interpretation.