
As the Senior Product Manager at DNASTAR, Matt Keyser works with scientists, software developers and support staff to create sequence analysis software that meets the current needs of researchers and that is ready to support future challenges and changing technology. In his past 18 years at DNASTAR, Matt has advised numerous customers on a wide array of sequencing and analysis projects, giving him a unique understanding of the challenges faced by scientists today.
One product that Matt has overseen at DNASTAR is the Variant Annotation Database, part of Lasergene Genomics. We recently sat down with Matt to ask why variant annotation is important. We also asked him a question that we commonly hear from customers: Once you find thousands of variants in a set of samples, how do you identify which ones are of the greatest importance?
Why is variant annotation important?
Identifying variants and annotating them with all that is known about them is important for interpreting their impacts on heredity, evolution and disease.
In many cases, the direct correlation between variation and disease is not obvious. Therefore, the effects on coding regions and protein structures are used to interpret the effects of the mutation in the living system.

Variant analysis can be used to:
Identify the cause(s) of disease by comparing the DNA of affected and unaffected individuals.
Understand evolution by doing phylogenetic comparisons of variants in different populations.
Identify somatic variations that occur in mixed cell populations of mosaic or tumor tissues.
Understand the biology of a mutation by learning how variants affect protein structure and function.
Identify variants in viral or bacterial strains that may affect the duration and intensity of an epidemic.
What type of sequencing data do I need for analyzing variants?
Some common sequencing technologies that create data suitable for variant annotation and analysis include Sanger/ABI, Illumina, Ion Torrent, PacBio and Oxford Nanopore. Sanger technology is still widely used for small scale variant analysis where accuracy is most important, while the Illumina platform provides both accuracy and high throughput variant analysis. Long read platforms have much improved accuracy for variant analysis and the extended read lengths also make additional analysis such has haplotype phasing and large structural variant analysis more practical.
During NGS sequencing for Illumina and Ion Torrent, the pipeline tools associated with the sequencing instrument usually clean up the data files. This is normally sufficient, but some output sequence files can benefit from scanning with a third-party tool like FastQC.
By contrast, Sanger data usually contains many base calling errors at the 5’ and 3’ ends where the chromatogram peaks are not high quality. This type of data requires a high-quality software program that can accurately trim the sequence ends prior to assembly.
What should I look for in a variant analysis software package?
To analyze variant data, you will first need software to align the experimental sequences against a known reference sequence. Reference sequences for common model organisms are available for free download at sites like NCBI.

Freeware tools are available for sequence assembly, but they are often difficult to use and require proficiency in several unrelated software components. Some of these tools also require the ability to use scripting languages.
Instead, look for assembly software that streamlines the entire workflow with minimal user interaction. For example, if your starting point includes raw sequence data and a reference genome, good software should be able to align data, detect variants, filter low quality variations, detect non-synonymous variants and then import additional variant annotations from a wide range of variant databases.
Why is the filtering step important?
Identifying more interesting variations from thousands of located variants can be challenging and often requires several rounds of analysis and data filtering.
Most variants are benign mutations in the DNA that do not affect protein coding and are not located in or near genes. Default filters should be in place to remove these variations from the initial analysis. Variant filtering can quickly eliminate thousands of variants from consideration and save you time and frustration.
What are some of the biggest challenges in variant annotation?
Most researchers begin by using existing annotation from the reference genome to identify variants in coding regions whose presence affects protein coding (AKA “non-synonymous” variants). Some organisms may also have sets of previously identified variants, usually in VCF file format. This annotation information can be imported into the analysis so the researcher can differentiate between previously characterized variants and novel variations.
Annotations for known variants may include information on allele frequency distribution, as well as references or links to additional annotation databases that can be used to interpret the functional impact of the variation.
Online variant annotation databases contain massive amounts of useful information but accessing this data can be challenging. Most databases are prohibitively large for manual searches. In addition, many use proprietary formats that require researchers to pay for access. For these reasons, incorporating variant annotations from multiple sources can be extremely unwieldy and time-consuming for researchers working with even small data sets.
Even with initial filtering, there can be thousands of variants remaining. The ability to apply additional filtering on the imported variant database annotations is critical to creating a manageable and meaningful data set for interpretation.
In a human data set, you could use the variant annotation information in the dbNSFP database to filter variants based on clinical, functional, or evolutionary conservation using a myriad of different annotation criteria. Variant annotation can also be imported via VCF files that can contain a wide range of different variant annotations.
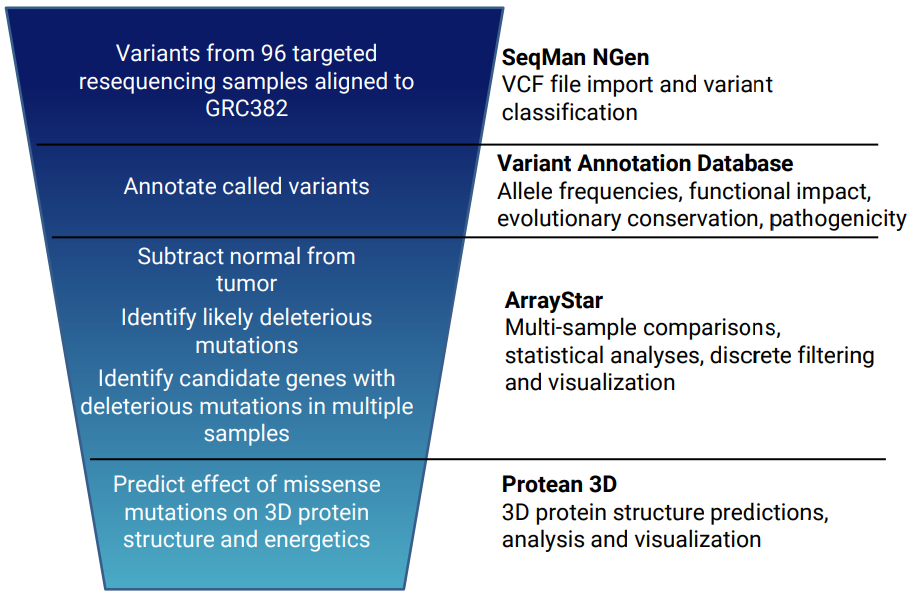
For example, we used progressive filtering (see image) to reduce a set of thousands of variants from 96 cancer patients to identify a small subset of genes with variants that are predicted to inactivate the gene in multiple tumor samples. Click the red button below to view a poster describing this experiment.
How much should I expect to pay for comprehensive variant annotation software?
Commercial software packages can cost a few thousand dollar or more per year, where academic and government users are typically eligible for discounted pricing. You can see pricing for Lasergene Genomics here.
There are also many open-source or free packages that can do parts of the variant analysis workflow. It is important to bear in mind that with free options, you will generally spend more time configuring workflows and moving data from one tool to another, and there may be limited support resources to guide you. Depending on available resources in your lab, the time and personnel expenses required to run open-source tools may exceed the costs involved in using commercial software.
What are the steps for the variant annotation workflow in Lasergene Genomics?
Step 1: Use SeqMan NGen to assemble sequences and detect variants.
Launch SeqMan NGen and select one of the “variant analysis” workflows (Figure 1).

Figure 1. The SeqMan NGen “Workflow” screen provides a variety of options for variant analysis based on data type and experimental goal.
Next, follow the wizard prompts to upload the sample sequences, reference sequence, and (optionally) .BED or .VCF files. Among other options, the wizard allows you to use one of DNASTAR’s curated genome packages as the reference. These packages are provided for most common model organisms. (Figure 2).

Figure 2. If you don’t already have a reference sequence, the SeqMan NGen wizard provides the option of downloading and using a genome package from the DNASTAR website.
If you are working with human data, be sure to check “Import Variant Annotation Database” from the Analysis Options wizard screen. This will provide extra annotation-related functionality when performing downstream analysis in ArrayStar (see next section).
Once you have uploaded your data and set up assembly options as desired, the wizard prompts you to begin assembly on your local computer or on the cloud, depending on the project size and your machine’s computing capacity.
When assembly finishes, SeqMan NGen provides buttons allowing you to perform downstream analysis in ArrayStar and/or in SeqMan Ultra. The option you choose depends on your experimental goals.
Step 2 (Option A): Use ArrayStar to identify, annotate, and filter variants.
Once the assembly opens in ArrayStar, a good first step is to filter the set to show only the variants of most interest to your research. Choose Filter > Filter All to use the advanced filter to locate variants using any combination of dozens of filtering options (Figure 3).

Figure 3. ArrayStar’s “advanced filtering” utility with several types of filters applied.
For example, you might elect to search for statistically significant, non-synonymous variants that only appear in certain samples and are already known to be deleterious. Many additional filters are available if you are working with human samples and checked the “Import Variant Annotation Database” box during setup in SeqMan NGen (see Step 1).
After applying filters, open the customizable SNP table to see the filtered variants; the ones likely to be of most interest to your research. Click any link in this table (Figure 4) to see the relevant online database entry for that variant.

Figure 4. ArrayStar’s SNP table with links to online database entries for each variant.
Step 2 (option B): Use SeqMan Ultra filter variants and view them within the alignment.
When you open the assembly in SeqMan Ultra, press the Show table of variants tool to show the Variants view (Figure 5).

Figure 5. SeqMan Ultra’s Variants view shows information about each variant, including location, impact, and significance.
A good next step is to click the Filter all variant tables tool and filter the table to only show variants of interest to you. For instance, you might require that the variant be non-synonymous, be a substitution (or an indel), meet certain significance or depth thresholds, or be present in specified databases.
Once only the variants of interest are displayed, double-click on any row to open the Alignment view, where you can see any the variant in the context (Figure 6).

Figure 6. SeqMan Ultra’s Alignment view lets you see each variant in the context of the entire contig.
Try Lasergene’s variant analysis workflow yourself
If you have sequencing or variant data already and want to try this workflow for yourself, you can get a 14-day fully functional trial of Lasergene, which includes all of the applications discussed in this article.
These step-by-step tutorials come with downloadable data and are a great way to get started with your free trial:



Leave a Reply
Your email is safe with us.