
The Analysis Options wizard screen allows you to specify the analysis parameters to use for your assembly. This screen comes in several variations, one of which is shown below:
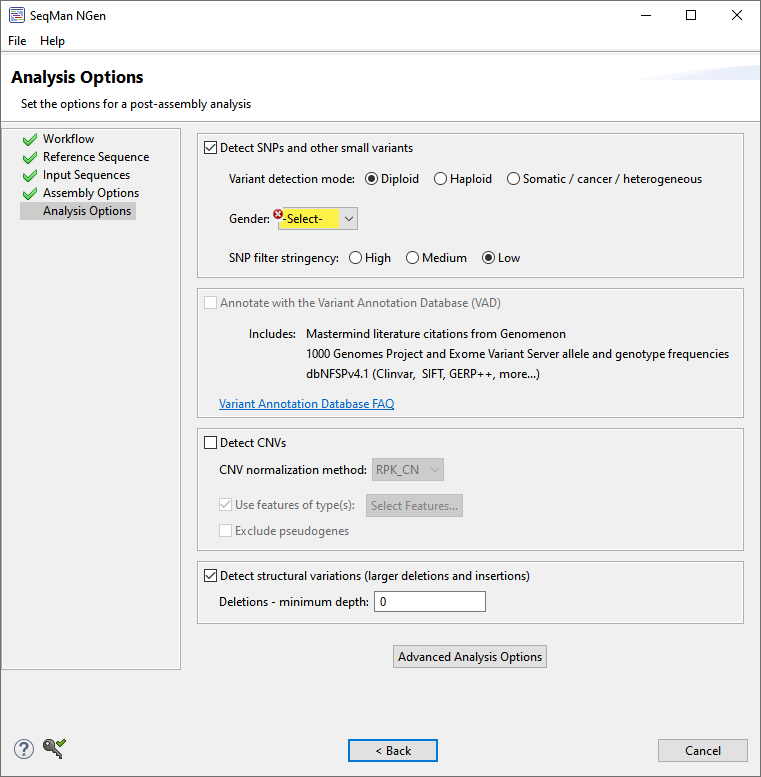
Depending on the workflow, only a subset of the following options will be available:
| Category | Options and Descriptions |
|---|---|
| SNP detection | To enable SNP detection, start by checking Detect SNPs and other small variants. If this box is checked, you can later open the assembly in ArrayStar or SeqMan Pro to view the SNPs. Use Variant detection mode to specify genome ploidy for SNP detection purposes. Choosing Haploid or Diploid establishes the statistical model SeqMan NGen will use in estimating the probability that a given called variant is real (i.e., that the sequence really differs from the reference). Selecting Somatic/cancer/heterogeneous (e.g. for a polyploid genome, cancer panel, etc.) prevents SeqMan from calculating probabilities. If the Gender checkbox is present, specify the gender of the subject (Male/Female), if known. Otherwise, select Unknown. This checkbox appears only if you are using a DNASTAR genome template package and have chosen a genome ploidy other than Haploid. SNP filter stringency specifies two key settings for placing a read in the layout. When building an assembly, SeqMan NGen uses a three stage strategy: overlap, layout, and alignment. In the overlap stage of a reference-guided assembly, for example, each read and the reference are broken up into an overlapping set of substrings or “mers” of a specified length (“mer length” or “mer size”). Identical mer matches are an indication that the read matches the reference at that position. The more overlapping mers between two sequences, the stronger the indication that the match is real. The layout stage uses that overlap information and attempts to place each read in its true position on the reference. The final layout of all the reads is then sent to an aligner that produces the final fully gapped alignment. Layout stringency settings can be used to adjust the extent of overlap data required to include a putative match in the final layout. The radio buttons specify stringency levels for “soft” filtering of SNPs. Soft filtering means that SNPs of the least interest to you will be automatically hidden when SNP reports/tables are viewed in SeqMan Pro, SeqMan Ultra or ArrayStar. Your selection in this screen controls the three assembly parameters shown in the table below. For more information on PnotRef, see Filter based on P not Ref.
|
| Variant Annotation Database | Check Annotate with Variant Annotation Database (VAD) if you are working with human samples and would like to import variant annotations from a specific portion of the NCBI RefSeq database maintained on the DNASTAR website. This checkbox is only available for human samples assembled against builds 37 or 38. This is an extremely powerful analysis tool that we highly recommend using any type you are analyzing human samples. To learn more about the Variant Annotation Database, click on the link in this section to read our Frequently Asked Questions (FAQ). |
| CNV detection | Check Detect CNVs if you wish to calculate copy number variants (CNV) as part of the assembly. If the box is checked, you may choose between two CNV Normalization method options: RPK-CN and None (i.e., no data normalization). If you also select a Variant detection mode other than Do not calculate variants, then CNVs, SNPs and small indels will be calculated from the assembly. After assembly, you can then use ArrayStar to view all three types, or SeqMan Pro or SeqMan Ultra to view only the SNPs and small indels. Check Use features of type(s) to only report results when a specific type of feature is used as the target for mapping reads. Note that mapping occurs regardless of the type of feature annotation. However, when you check this option, the mapping results for unwanted feature types will not be reported. Put checkmarks next to the feature types you wish to use, then press OK.  If you leave Use features of type(s) unchecked or if the reference sequence has no feature annotations, each individual sequence in the reference set will be used as a separate transcript (i.e., a single gene feature). Check Exclude pseudogenes to not report mapping results for features with /pseudo in their annotations. As with the previous option, mapping occurs regardless of the type of feature annotation. |
| RNA-Seq normalization | Check Normalize RNA-Seq values if you want to apply a normalization method to the data on a per-isoform basis. If you check this option, use the drop-down menu to choose the desired RNA-Seq normalization method. In order to enable this option, some workflows require you to check the Calculate Copy Number Variation option. See CNV detection, above, to learn about the Use features of type(s) and Exclude pseudogenes options. |
| ChIP-Seq normalization | Check Set ChIP-seq peak detection method if you want to apply a normalization method to the data. If you check this option, use the drop-down menu to choose the desired ChIP-seq peak detection method. See CNV detection, above, to learn about the Use features of type(s) and Exclude pseudogenes options. |
| Structural variations | If you want SeqMan NGen to flag structural variations for viewing during downstream analysis, check the box next to Detect structural variations (larger deletions and insertions). If desired, type in a minimum depth to be considered a deletion. Note that structural variation options are not present in this dialog when following a long read workflow. |
To access additional options, click the Advanced Analysis Options button to open a multi-tabbed dialog. Each tab has changeable parameters for different parts of the analysis process. Different workflows have different subsets of tabs. In addition, tabs with the same names may contain different options depending on the workflow. For details on each tab, see Peak Detection tab, Variants tab and Layout tab.
Click Next > to proceed to the next wizard screen or < Back to return to the previous screen.
Need more help with this?
Contact DNASTAR