
This wizard screen only appears for reference-guided ChIP-seq workflows. The Define Binding Protein screen allows you to define binding sites for your experiment.
Start by making a selection from the Known binding site motif drop-down menu. The Define Binding Proteins screen will change and offer different options based on your selection.
| Unknown |  Choose this option to use the whole genome to find peaks. Type a name into the Binding Protein Label text box. |
|---|---|
| Type-in pattern |  Select this option to specify your own binding site pattern for your binding protein. Type a pattern into the Binding Site Sequence text box and a name into the Binding Protein Label text box. SeqMan NGen recognizes IUPAC nucleic acid and regular expression syntax for these patterns. A key to the syntax is provided within the wizard screen. |
| Transcription Factor database | 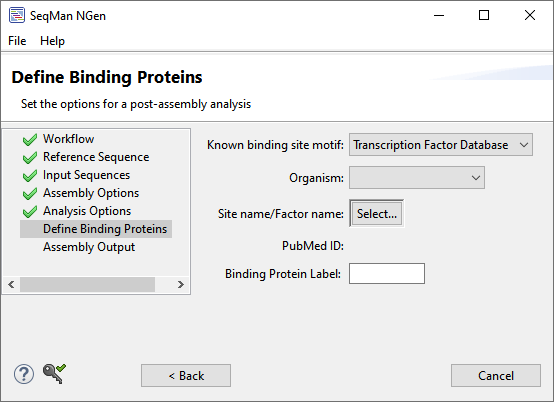 Choose this option to select a binding site pattern from DNASTAR’s transcription factor database, which is for prokaryotic organisms only. Select the Organism from the drop-down menu. Click the Select button to choose the site/factor name from a list. Make a selection and choose OK. The PubMed ID field will be filled in automatically. Finally, type a name into the Binding Protein Label text box. |
| JASPAR (PWM) |  Select this option if you want to use the JASPAR position weight matrix to locate binding sites for eukaryotic organisms. If you choose this option, SeqMan NGen will calculate the log-odds for each sequence given the selected matrix. The score for a single character at a particular position in the matrix is equal to the log2 of the likelihood of seeing that character at that position in the data used to generate the matrix divided by the background likelihood of seeing that character at that position. For example, if the matrix is derived from 80 sequences and in 70 of those sequences there is an “A” in position 1, the log odds score of seeing the character “A” in position 1 is log2((70/80)/(20/80)) = 1.80. If a “C” occurs 1 time in position 1 of the training sequences, the log odds score of seeing the character “C” in position 1 is log2((1/80)/(20/80)) = -4.32. To get the log odds score for the whole sequence, SeqMan NGen sums the log odds scores of each character in the sequence. A sequence is considered to "match" the matrix if its score is greater than or equal to the specified Threshold. By default the threshold value is half of the average of the log-odds scores of sequences that were used to train the pattern. You can increase the threshold for more stringency or decrease it for more matches. Once you initiate sequence assembly, all detected peaks will be scanned for the presence of sites that pass the JASPAR scoring threshold. Once you have chosen this option, select the Organism from the drop-down menu. Click the Select button to choose the site/factor name from a list. Make a selection and choose OK. The remaining fields will be filled in automatically. If you wish to view online entries for the selected site/factor, click on the corresponding links. |
Click Next > to proceed to the next wizard screen or < Back to return to the previous screen.
Need more help with this?
Contact DNASTAR