
After running the long read assembly in Part A, the following steps can be used to evaluate the base level accuracy of the assembled sequence. The Quality Assessment Tool for Genome Assemblies (QUAST) is utilized to compare the assembled consensus sequence to the E. coli reference sequence.
Before you begin, you will need to decide whether you want to use the QUAST Web Interface or download and install the local version of QUAST. In order to use the local version of QUAST, Python must be installed on your computer before you begin.
Both the reference sequence and the assembly output must be in .fas or .fasta format. The sequences in this case are:
- Assembled sequence: This is the exported consensus from the completed assembly from Part A. It can be found in the Project Folder you specified during assembly setup in the Additional output subfolder. The file name will end in longReadAssembly_consensusSeqs.fas.
- E. coli reference sequence: U00096.3.fas.
To perform the evaluation using the QUAST web interface:
- Go to the QUAST Web Interface.
- Under Assemblies, upload the sequence file ending in longReadAssembly_consensusSeqs.fas.
- Under Genome, click Another genome and upload U00096.3.fas as the Reference. Enter a Name for the genome.
- Click Evaluate.
When analysis is complete, the results will appear in the browser window. QUAST provides an option to enter your email address so that you can return to the page later.
To perform the evaluation using a locally-installed version of QUAST:
- Download and install the local version of QUAST (you must already have Python installed).
- Launch the Command Line (Win) or Terminal (Mac).
- Type the following text: >[path]\quast.py [path]\[project name]_longReadAssembly_consensusSeqs.fas -R [path]\U00096.3.fas. Press Enter.
When analysis is complete, the results are saved by default in the Users directory in a quast_results folder.
Interpreting QUAST results:
QUAST produces a number of useful output files for evaluation.
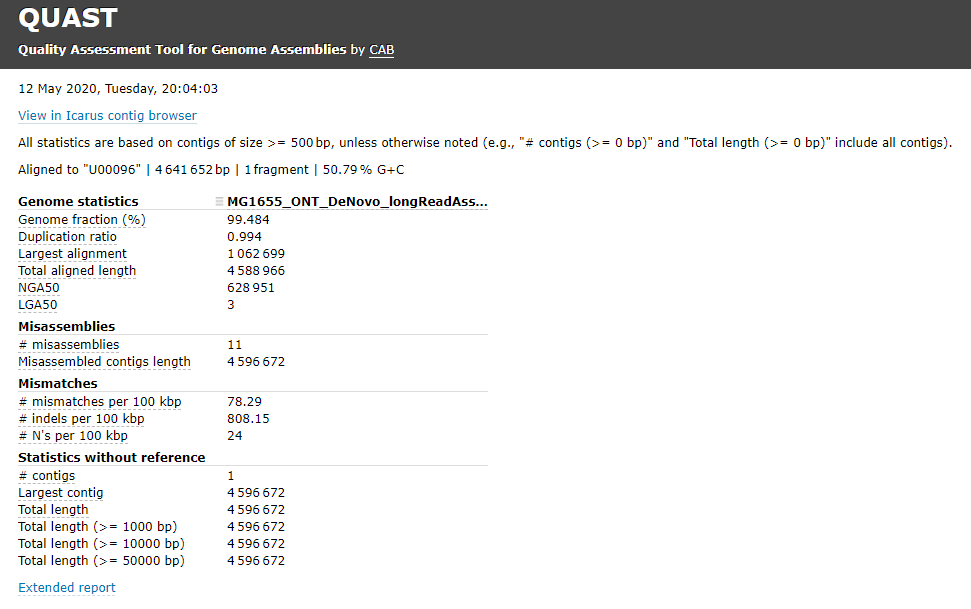
To calculate a first approximation base-level percent accuracy value for the consensus sequence:
- Open the report.pdf file.
- On the Report page find the Total aligned length.
- On the Misassemblies Report page, find the entries # Mismatches and Indels length.
- Accuracy = 100 * (Total aligned length – # Mismatches – Indels length) / Total aligned length
The initial SeqMan NGen beta version of the long-read assembler typically produces a single full length contig of the MG1655 genome with an approximate base-level percent accuracy of 98.65%. The total length of the consensus sequence may be shorter than the expected reference sequence value of 4,641,652 largely due to the number of “deletions” caused by errors in data as well as outstanding issues in the aligner of this beta release.
This marks the end of this tutorial.
Need more help with this?
Contact DNASTAR