
In Part A, you will use SeqMan NGen to run the assembly, then launch the results in SeqMan Ultra.
- Download T5_Long_Read.zip (232 MB) and extract it to any convenient location (i.e., your desktop). Part A of the tutorial will use the files MAP006-1_2D_pass.fastq (sample) and U00096.3.gbk (ref).
- Launch the workflow in the SeqMan NGen wizard using either of the methods below:
- Launch the DNASTAR Navigator, click on the Genomics tab on the left. On the right, click De novo genome assembly and editing. This launches SeqMan NGen at the Workflow screen. On the right, under PacBio/Nanopore options, click De novo.
- Launch SeqMan NGen and choose New Assembly. In the Workflow screen, choose De Novo Genome Assembly and Editing on the left. On the right, under PacBio/Nanopore options, click De novo.
- Launch the DNASTAR Navigator, click on the Genomics tab on the left. On the right, click De novo genome assembly and editing. This launches SeqMan NGen at the Workflow screen. On the right, under PacBio/Nanopore options, click De novo.
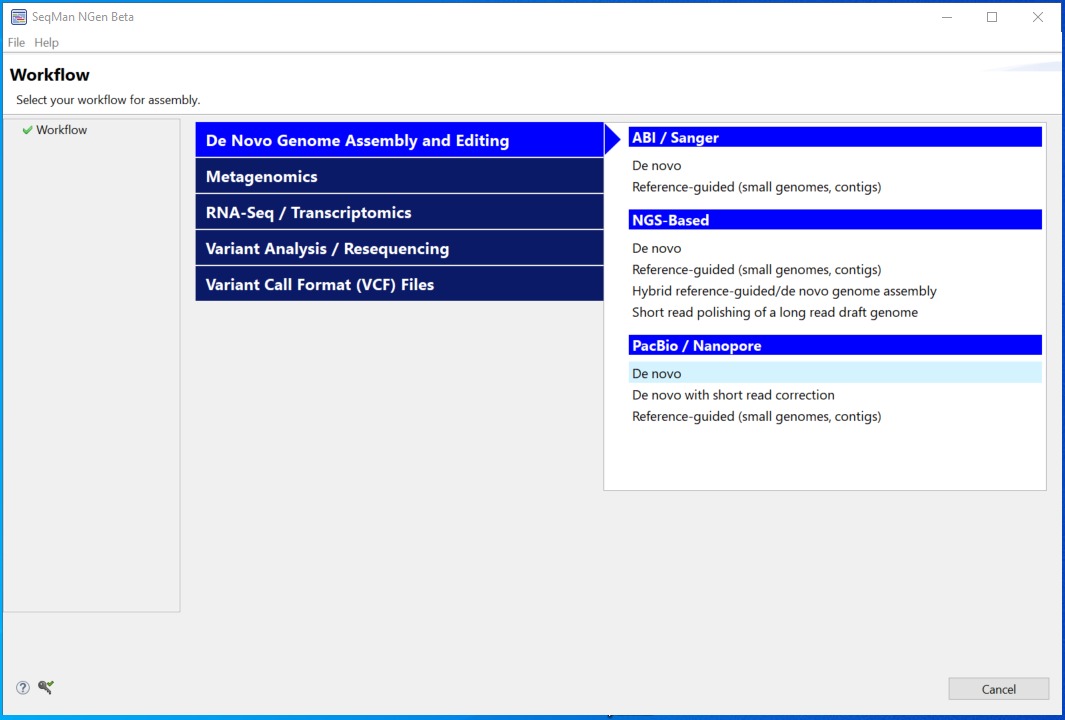
- In the Input Sequences screen, choose a Read Technology of ONT. Click the Add button and add the sequence MAP006-1_2D_pass.fastq.
Click Next.
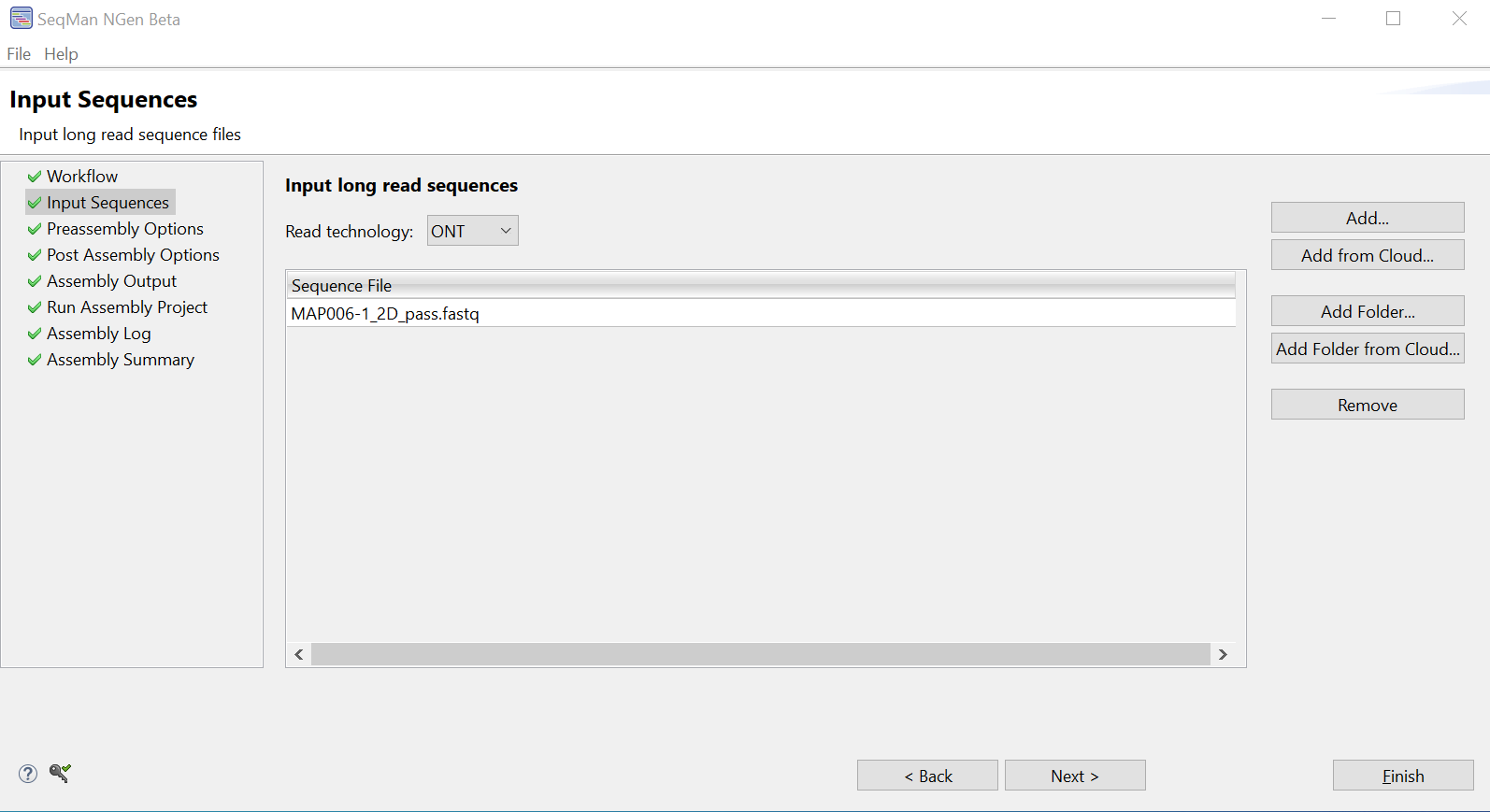
- In the Preassembly Options screen, enter an Expected genome length of 4600000 (46 followed by 5 zeroes). Leave the Desired depth of coverage of final assembly at the default setting of 100. Leave Use longest reads in data set to achieve depth selected.
Click Next.
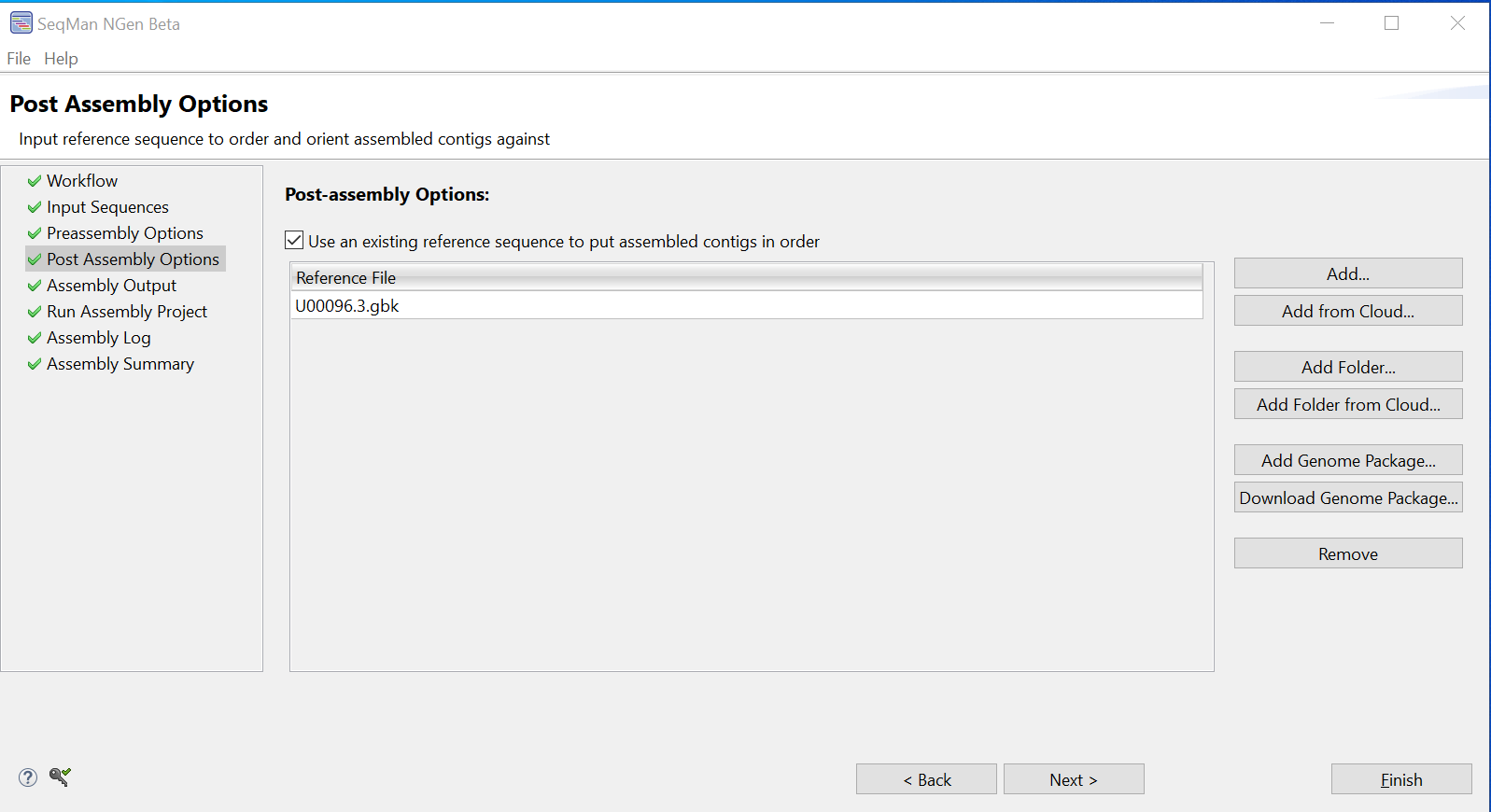
- In the Post Assembly Options screen, keep the box checked and use the Add button to add the reference genome U00096.3.gbk.
Click Next.
- In the Assembly Output screen, type in a Project Name. Then use the Browse button to select a save location for the project. Click Next.
- In the Run Assembly Project screen, look at the “Recommended” method under Run assembly.
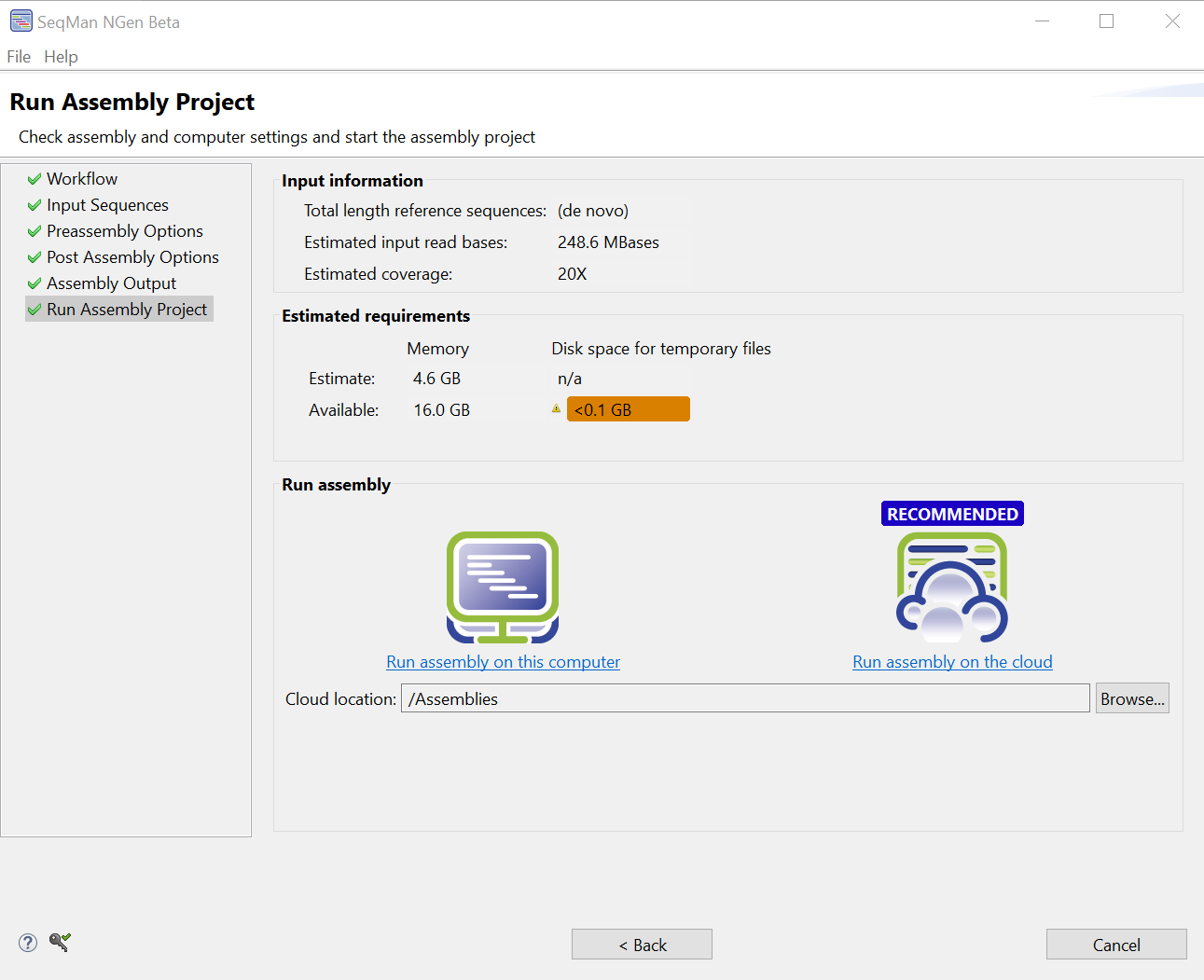
Regardless of the recommendation, it is probably safe to choose Run assembly on this computer (see Note below). Typically, local assembly takes 30-60 minutes.
- A message should appear at the bottom of the Assembly Log screen indicating that the assembly has finished successfully.
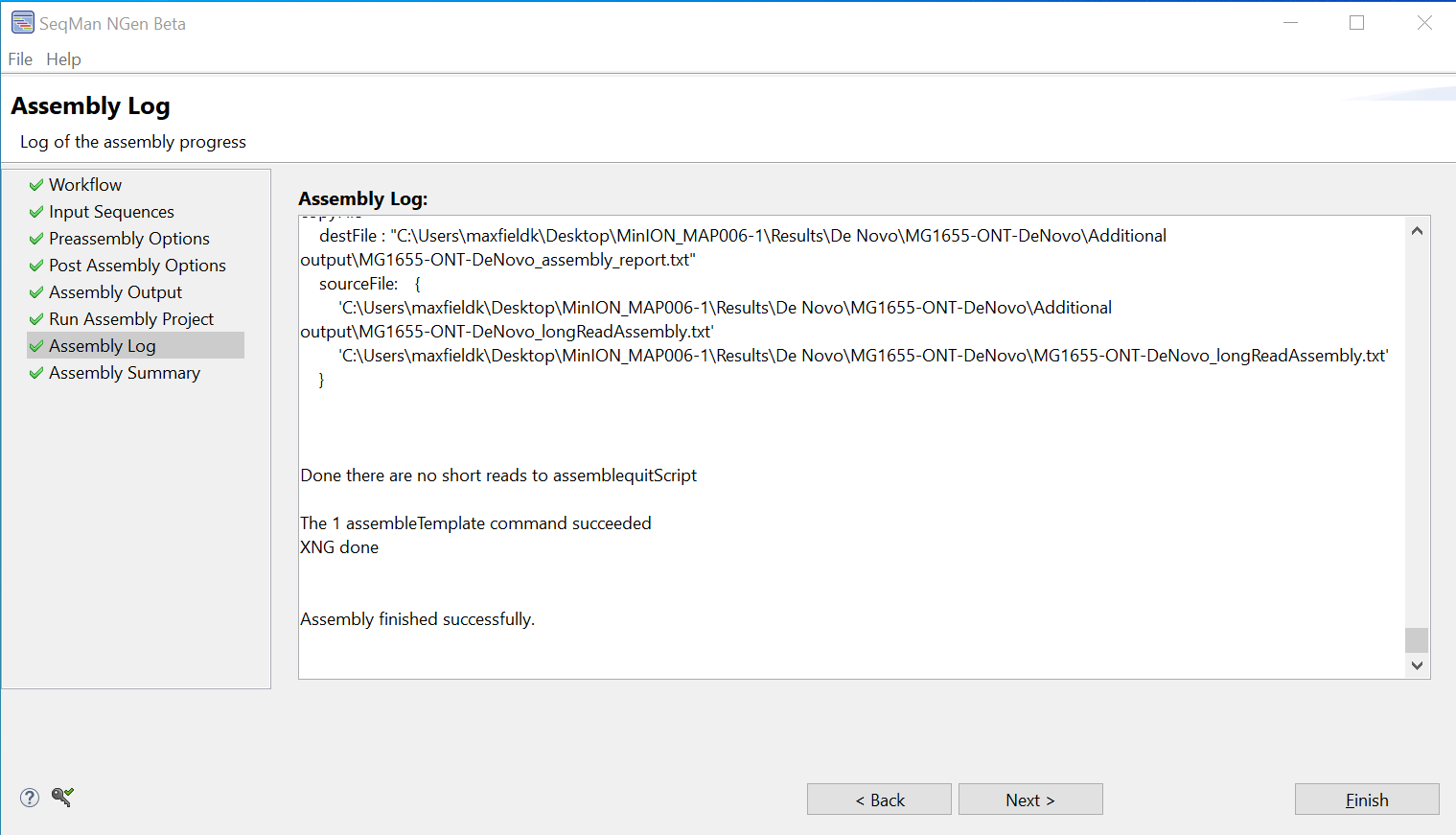
Click Next.
- By scrolling through the bottom part of the Assembly Summary screen, you will see it consists of three sections:
- Run statistics is a high-level summary that includes the number of input and assembled reads and the assembly time.
- Script displays a portion of the run script and contains useful debugging information.
- Contig/Scaffold statistical summary includes information on the number of contigs assembled, their length and average depth of coverage. Note that the contig N50 value is includes consensus gaps.
- Run statistics is a high-level summary that includes the number of input and assembled reads and the assembly time.
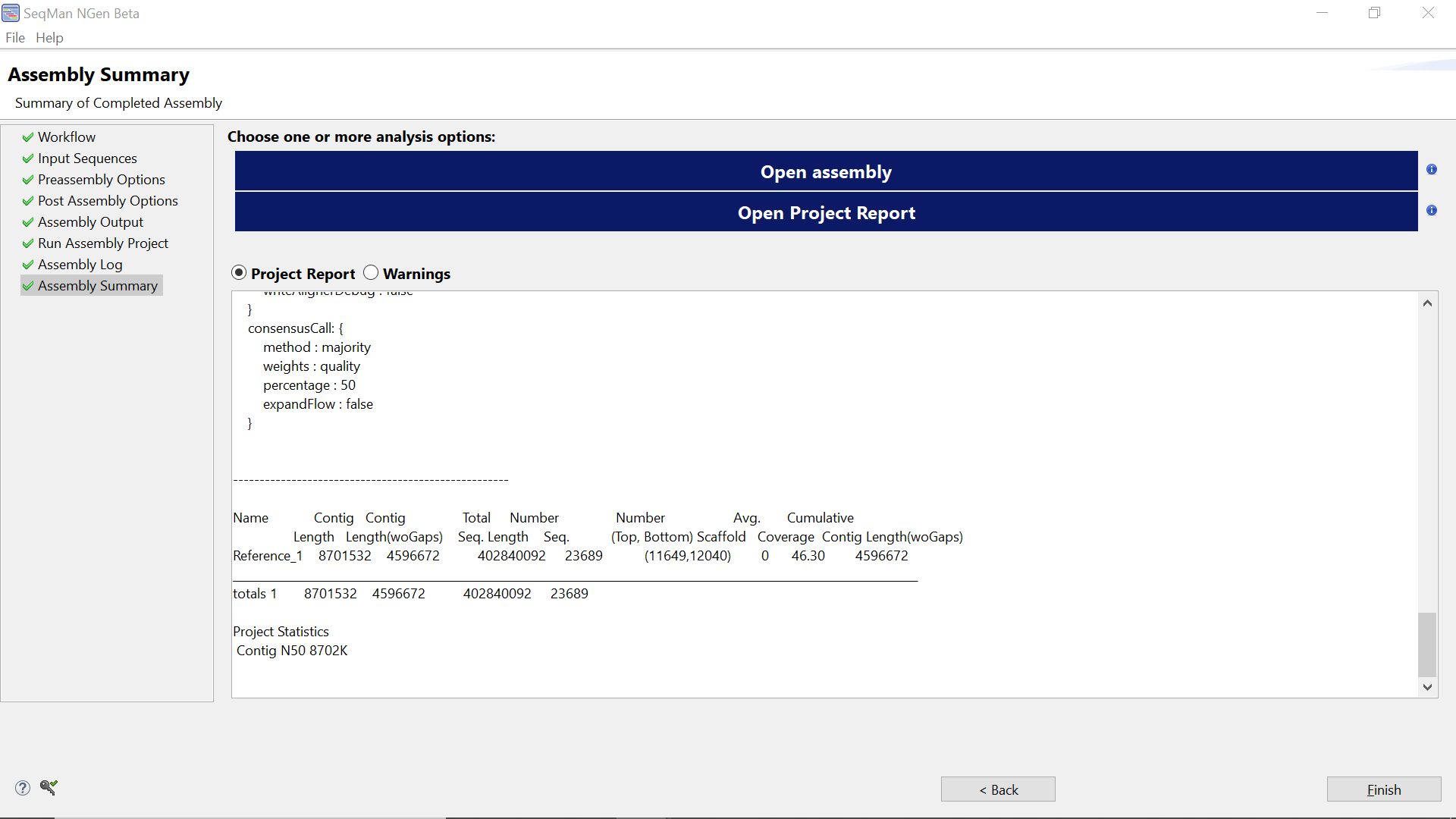
As shown in the image above, the single contig produced by this assembly may be somewhat shorter (e.g., ~4.60Mb) than the complete_E. coli_ genome (~4.64Mb) . This is due to a number of bases being erroneously “deleted”.
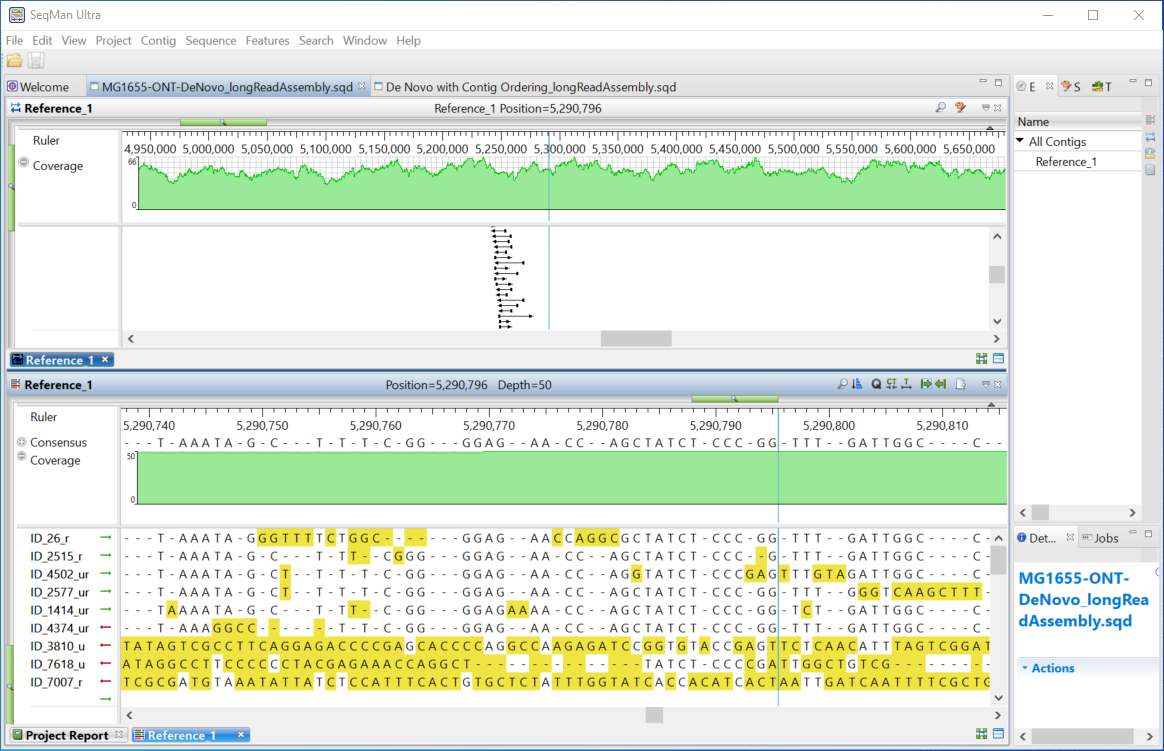
- Click the blue Open assembly button to launch assembly results in SeqMan Ultra.
- In SeqMan Ultra, open the Alignment and Strategy views and scroll to inspect the assembly. You will notice a significant number of gaps in the alignment caused by the relatively high error rate of the reads.
If desired, proceed to Part B: Evaluating assembly accuracy using QUAST.
Need more help with this?
Contact DNASTAR