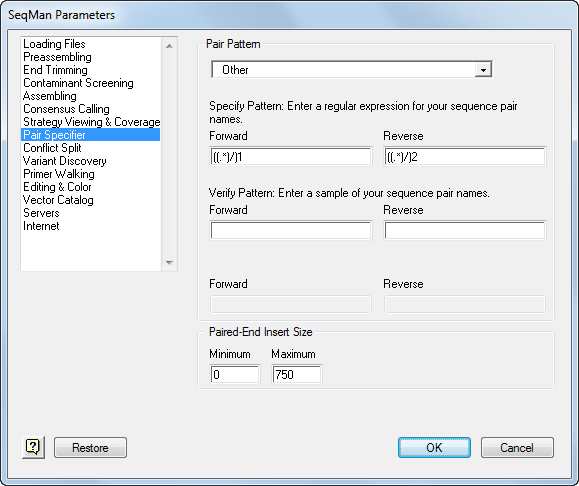
The Pair Specifier parameters allow you specify distinct naming patterns for forward and reverse sequence reads when assembling paired end data. Access these parameters by selecting Project > Parameters and choosing Pair Specifier from the list on the left.
The Pair Pattern drop-down menu allows you to select from among predefined patterns, Other (user-defined patterns), and No Pairs. Select No Pairs if you are not using paired-end data. It doesn’t matter which direction you call “forward” and which you call “reverse” in orientation, because SeqMan Pro evaluates both orientations for every read when assembling sequences. For paired end sequencing, the important point about any pair is not their absolute orientation, but rather that the two reads are of opposite orientation. Orientation is not considered in variant discovery.
If your pair-naming convention matches one of the predefined pair patterns, select it from the list:
•sample_f.abi < > sample_r.abi
•sample100.f_abc.abi < > sample100.r_abc.abi
•sample_n100.abi < > sample_f100.abi
•SAMPL0D1234.abi < > SAMPL0E1234.abi
•sample#001/1 < > sample#001/2
•ID_1234_f < > ID_1234_r
If none of the predefined pair patterns matches your pair-naming convention, select Other. Then, in the Specify Pattern section, type regular expressions to match your forward-reverse pair pattern using SeqMan Pro’s Paired End Specification Language.
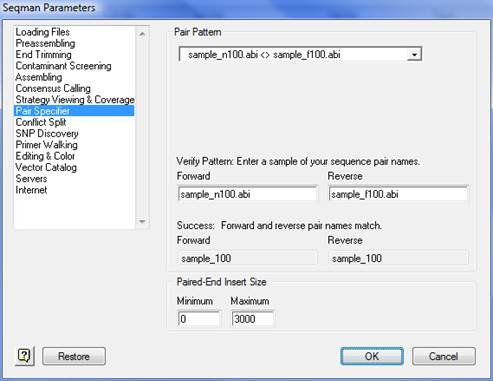
In the Verify Pattern section, type a sample of your sequence names in the Forward and Reverse fields.
•A ‘Success’ message indicates that your names match the pattern you chose. The common parts of the name are listed in Forward and Reverse fields.
•An ‘Error’ message indicates that our names do not match the pattern you chose. Choose another pattern or define your own by choosing Other in the dropdown menu.
In the Paired-End Insert Size section, you can specify minimum and maximum values (in bases) for the paired end insert size. Depending on your assessment of the potential for experimental error, you may want to make this size range bigger than the size range targeted in your sequencing projects. Be aware, however, that making the size range too large can cause false joins.
Note: When size data are used for contig ordering, the sizes correspond to the length of the inserts, not the gap remaining between the 3’ends of paired reads extending into the inserts.