Note: This topic is not applicable to BAM-based projects.
Consensus Calling parameters control how the consensus sequence is determined.
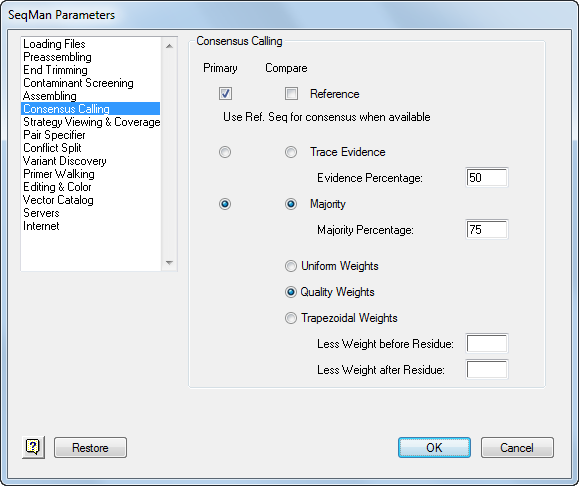
Access these parameters by selecting Project > Parameters and choosing Consensus Calling from the list on the left.
•Using the radio buttons, choose one method to be the Primary consensus calling method and another to be the Compare method. The Alignment View allows you to see a comparison of consensus sequences calculated using both methods.
Note: If you import an assembly into SeqMan Pro, the Imported consensus is used as the default Primary method.
•Select whether to use Trace Evidence or a simple Majority. Choose Trace Evidence if fluorescence trace data are available, or Majority if your data consists of text sequences.
o Trace Evidence directly reflects the quality of peaks in the trace data and is the most accurate method for generating consensus sequences for contigs. It is strongly recommended you use Trace Evidence for consensus calling whenever trace data are available. Sharp, well-defined peaks are assigned higher scores than less well-defined peaks. For more information, see Quality Score Calculations.
o The Evidence Percentage value controls the stringency used by the Trace Quality Evaluation system to make unambiguous calls in the consensus sequence. The default setting for Evidence Percentage was chosen as the value that yielded the highest consensus accuracy among a test set of fluorescence trace data obtained from a genome sequencing center. You may want to adjust this value for data sets of different quality, or when you expect to find heterozygous positions in sequence reads. For more information, see Controlling Ambiguity Calling.
o Majority lets you set the Majority Percentage: the minimum percentage of identical residues at each position required to identify the consensus base at this position. Ambiguous bases in the consensus denote a column of bases whose constituents do not meet this threshold. Ambiguous codes are assigned in these cases.
•Uniform Weights, when checked, makes every residue equal in scoring value for an assembly.
•Quality Weights can be applied when constituent sequences are derived from chromatogram files. The bases called by the base caller are weighted according to the quality of the underlying sequence trace data, so that poorer quality data are de-emphasized in calling the consensus. This method can yield superior results to uniform or trapezoidal weighting but is usually not as accurate as using Trace Evidence, because Trace Evidence adjusts the quality weights taking into account competing evidence for other bases in each peak region of the chromatogram.
•Trapezoidal Weights improves the accuracy of the consensus by de-emphasizing data toward the 5’ and 3’ ends of gel reads, on the assumption that the terminal portions of sequence reads are less reliable than the middle regions.
o Less Weight before Residue reduces the weight used to calculate the consensus sequence for bases before the indicated position, for each constituent sequence in a project.
o Less Weight after Residue reduces the weight used to calculate the consensus sequence for bases after the indicated position, for each constituent sequence in a project.