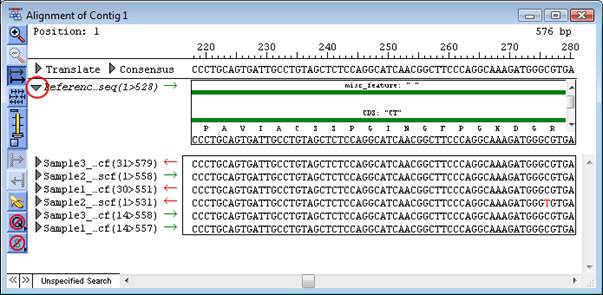
If a .seq, .sbd or .bam file has been annotated with features prior to assembly, the features can be displayed by clicking the triangle to the left of the sequence name in the Alignment View.
Features are drawn directly above the sequence they are associated with.
If a sequence in your project has many overlapping features, use the Amplitude Control Tool from the Alignment View toolbar to increase the vertical space allotted for features. If necessary, use the vertical scroll bar found to the right of the feature area to scroll through the features for your sequence.
All features appear as green or red arrows, with the translation, if applicable, displayed beneath it. (The green and red colors represent opposing orientation.) Translations are displayed for all exon and CDS features, as well as any feature that contains a /translation qualifier. The /codon_start qualifier for the feature is used to determine the start of the translation.
Above the arrow, the feature type and name are displayed. The /dnas_title qualifier is used for the feature name. If no /dnas_title qualifier is available, SeqMan Pro will use the value of the first qualifier listed for the name.
Hovering your mouse over a feature will display its position as well as the first few qualifiers for that feature:

Note: Features of the type “variation” may also be displayed in the Alignment View. See Viewing Variants in the Alignment View for more information.
Inferred features:
An asterisk next to a feature name indicates that the feature was inferred by SeqMan NGen. For example, if you assembled data in SeqMan NGen using a template that contained a tRNA feature—without a corresponding gene feature—then "gene" would be inferred at that position and would be displayed as a feature in the view. However, the inferred gene would be marked with an asterisk, as below:
