
The Features view is one of the ten views available in the Document window, and shares a sub-set of button tools with the other views. To make it the active view (or one of two active views), click on the Features tab (  ).
).
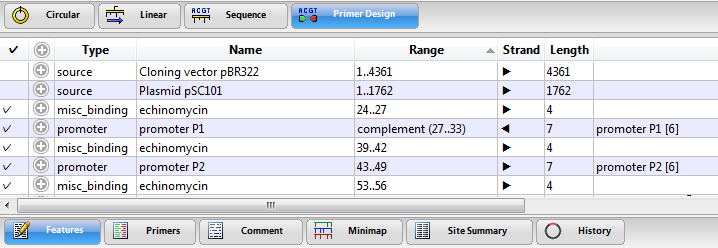
The Features view displays a table of features (annotations) that have previously been identified in the DNA or protein sequence. Features may be user-defined, or they may be part of the input file from GenBank or from other Lasergene applications. For information on creating, editing or deleting features, see Features.
The following table describes how to perform various tasks within this view:
| Task | How to… |
|---|---|
| To select one or more features in all graphical views | Single click on one feature row; or select multiple features using Ctrl/Cmd+click, Shift+click, or by dragging the mouse across the desired rows. Selected features are simultaneously selected in the Circular and Linear views. |
| To select the feature and its corresponding sequence, and to scroll to its location | Double-click on a compacted feature row or select the feature row and choose Feature > Select Sequence. |
| To expand/contract a feature row | Click a plus icon to expand the row and show details about the feature; click a minus icon to hide feature details. As a shortcut, you can use Ctrl/Cmd+Alt to toggle between expanding/contracting all rows. |
| To edit feature information | Expand a row, as described above. Insert your cursor in a field and type the desired information. |
| To sort data alphabetically within a column | Click on the column header. |
| To see the number of the features in total, the number visible, and the number selected | Look at the footer at the bottom left of the view.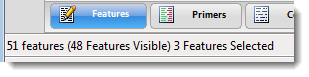 |
Columns in the Features view are described below:
| Column name | Description |
|---|---|
 |
Rows with a checkmark in this column denote a feature that is currently visible in the graphical views (Circular, Linear, Sequence). To hide a feature, click its checkmark. |
 / /  |
See “To expand/contract a feature row” in the table above. |
| Type | The feature type. |
| Name | The feature display name. |
| Range | The range of the sequence included in the feature. |
| Strand | The strand where the feature is located (forward  or reverse or reverse  ). ). |
| Length | The length of the feature in base pairs. |
| Description | Often the contents of the first /note qualifier. |
Need more help with this?
Contact DNASTAR