
The tools located in the upper right of the Alignment view are used to accomplish the following tasks.
| Task | How to |
|---|---|
| To display the search tools | To display the search tools in the upper left of the Alignment view, use the Find tool (  ). ). |
| To sort constituent sequences | To sort read files (the files in the left margin of the view) according to the selected criteria, use the Sort reads tool ( ). ).
|
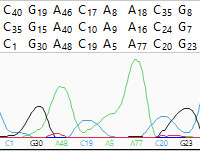
| To hide/show quality scores | Use the Quality scores tool ( ). This tool only functions when a) trace data was used in the assembly. ). This tool only functions when a) trace data was used in the assembly.
|
| To adjust the zoom level | Use the Compact text tool ( ) to adjust the zoom level to maximize the number of bases while keeping the text legible. Use the Restore default zoom tool ( ) to adjust the zoom level to maximize the number of bases while keeping the text legible. Use the Restore default zoom tool ( ) to adjust the zoom level to the (wider) default setting, making the text more comfortable to read. ) to adjust the zoom level to the (wider) default setting, making the text more comfortable to read. |
| See how the sequence is anchored | Prior to correcting an .sqd alignment with trace data (only), use the Anchor editing left (insert right) tool ( ) to see how the sequence is anchored. If the left anchor is active, residues left of the insertion point are held in place. If you insert any characters, the downstream sequence slides to the right. If you delete any bases, the downstream sequence fills in the gap by sliding left. Use the Anchor editing right (insert left) tool ( ) to see how the sequence is anchored. If the left anchor is active, residues left of the insertion point are held in place. If you insert any characters, the downstream sequence slides to the right. If you delete any bases, the downstream sequence fills in the gap by sliding left. Use the Anchor editing right (insert left) tool ( ) in a similar way. ) in a similar way. |
| Slide a Sanger sequence relative to other sequences | Use the Allow sliding sequences tool ( ). When this tool is activated, the cursor is shaped like a banana. Use your mouse and the banana-shaped cursor to drag any sequence in the Alignment view to the left or right, as desired. When you are done, click the Banana tool again to return the cursor to its normal state. Note that this tool is only active when the alignment contains Sanger trace files. ). When this tool is activated, the cursor is shaped like a banana. Use your mouse and the banana-shaped cursor to drag any sequence in the Alignment view to the left or right, as desired. When you are done, click the Banana tool again to return the cursor to its normal state. Note that this tool is only active when the alignment contains Sanger trace files. |
| To force SeqMan Ultra to call bases where ambiguity codes are currently displayed | Use the Show/hide variant degeneracy tool ( ). To restore display of ambiguity codes, press the tool again. ). To restore display of ambiguity codes, press the tool again. |
| Export data | Use the Export data tool (  ). This tool acts as a shortcut to the File > Export Data > Consensus command and opens the same popup dialog. See Export data to a file for information on using the dialog. ). This tool acts as a shortcut to the File > Export Data > Consensus command and opens the same popup dialog. See Export data to a file for information on using the dialog. |
| Export image | Use the Export image tool ( ). This tool acts as a shortcut to the File > Export Image > Alignment command. See Export an image of a view for more information. ). This tool acts as a shortcut to the File > Export Image > Alignment command. See Export an image of a view for more information. |
Need more help with this?
Contact DNASTAR