
The Add Features template, located in the Templates panel, lets you add specified features to the input sequences and saves the result. Alternatively, you can use this template to filter a variant call format file (.vcf) and save the results as a new VCF file.
Inputs consist of a sequence file (typically, one lacking features) and either a feature file (e.g., a .starff file) or a second sequence file that contains features. Note that if a second sequence is used, it can contain substitutions, compared to the sequence file, but may not have insertions or deletions.
The following examples illustrate some scenarios where this template may be useful:
- You want to add annotations to a sequence. With this template, you can define the annotations in a table of features (.starff feature file), add them to a sequence, and store the result. While annotation can be done manually in SeqBuilder Pro, SeqNinja’s Add Features template is useful for generating many features to a table format, then writing them to a sequence file format.
- You have two copies of the same biological sequence. One copy is a FASTA (.fasta) sequence with some capital letters that you wish to retain. The other copy is an annotated GenBank (.gbk) sequence. With Map Features, you can add the specified annotations from the GenBank file to the FASTA file, while still maintaining the capital letters in the latter.
- You have a large VCF file that contains extraneous data; for instance, data for all chromosomes when you are only interested in one particular chromosome. You can filter this large VCF file and save only the portion of it that corresponds to your area(s) of interest.
![]()
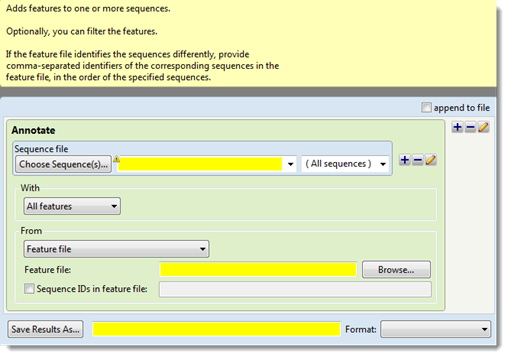
- In the “Sequence file” area, use the Choose Sequence(s) button to specify the input sequence file that you wish to annotate (see Add and modify a sequence).
- Use the “With” drop-down menu to select which features you want to add to that sequence: All features, Features matching or Features except. If you choose anything other than All features, the step expands to contain a new row.
![]()
In the new row, select the feature type of interest (gene, CDS, exon, intron, mRNA, tRNA, promoter or misc_binding) from the drop-down menu on the left, or type its name in the menu box. If you want to extract more than one feature type to the same output file, use the plus (+) button to add additional feature type lines. If you want to further limit the search to features matching (or not matching) particular qualifiers, check the Filter box, then choose a qualifier (gene, /product, /locus_tag, /note or /db_xref) from the drop-down menu to its right. In the right-most textbox, enter the text that the qualifier must match or not match (e.g., /gene = thrL) in order for the feature to be added to the sequence file. You may use wildcards in this box if you wish (e.g., /gene = thr*).

- Use the “From” drop-down menu to choose the type of file that will supply the annotations: Feature file or Same sequence(s) with different features.
- In the “Feature file” area, use the Browse button to navigate to the file.
- (optional) To allow a sequence ID in the feature file to differ from the ID in the sequence data, check the Sequence IDs in feature file box. Then use the text box on the right to specify comma-separated identifiers of corresponding sequences in the feature file. There is no need to reorder the IDs in the VCF file. Rather, the order of the identifiers must correspond to the order of sequences in the sequence data.
Example 1: You want to add VCF file features to a set of human chromosome sequences. However, the identifiers for chromosomes in the VCF file do not match the identifiers for the sequences. For example, a sequence might be identified in a GenBank file as NC_000022, but in a VCF file as 22. Or perhaps one sequence file uses the names chr1, chr2, chrM; and the other uses 1, 2, MT.
Example 2: The sequences are ordered numerically {NC_00001, NC_00002,…} but the VCF variants are ordered alphabetically {1,10,11,…,2,20,…}. There is no need to re-order the variants in the VCF file. Instead, enter the VCF sequence identifiers in the order of the sequences in the sequence file: { 1, 2, …, 10, 11, … }.
- In the Save Results As area, choose the name, location and format in which to save the output (see Specify output format and location). Choose the option “VCF” to write the output in variant call file format (.vcf).
Need more help with this?
Contact DNASTAR