
The Clustal Omega algorithm is for gene level alignment of either protein or nucleotide sequences. To run a Clustal W alignment, select two or more sequences and choose Align > (Re)Align Using Clustal Omega.
If you wish to change method options, instead choose Align > Align with Options. The dialog is the same whether the sequences are protein or nucleotide.
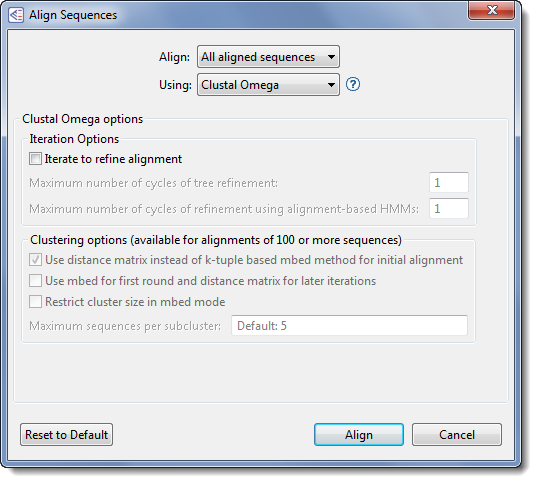
Change settings as desired:
- Use the Align drop-down menu to select sequences to align or realign.
- In the Using drop-down menu, choose Clustal Omega.
- To refine the alignment by specifying a threshold for iteration cycles, check the box next to Iterate to refine alignment. When this box is checked, two refinement options become available. You may enter values for neither, either, or both options.
- Enter a value for Maximum number of cycles of tree refinement to specify a cutoff for the maximum number of iterations. The default is 1.
- Enter a value for Maximum number of cycles of refinement using alignment-based HMMs to set a cutoff for the number of HMM iterations. MegAlign can often improve the placement of gaps by comparing the alignment to a Hidden Markov Model (HMM) of the alignment. The default is 1. The HMM is very slow, so be aware that entering a large number may cause a commensurate decrease in alignment speed.
- Enter a value for Maximum number of cycles of tree refinement to specify a cutoff for the maximum number of iterations. The default is 1.
In both cases, MegAlign Pro will recompute the pairwise distances, recalculate the guide-tree, and then realign the sequences while staying within the maximum cycle threshold.
- If your project contains ≥ 100 sequences, three clustering options are available. All three options are disabled for projects containing fewer than 100 sequences.
- To force clustering using a distance matrix based comparison, check Use distance matrix instead of k-tuple based mbed method for initial alignment.
- To use mbed for the first iteration and a distance matrix for other iterations, check Use mbed for first round and distance matrix for later iterations.
- If you check the second box, but not the first, you may optionally set a threshold for the cluster size during mbed iterations by checking Restrict cluster size in mbed mode. Once you have checked that box, you may elect to over-ride the default by entering a value for Maximum sequences per subcluster. The default is <number of sequences to be aligned> minus ‘1,’ up to a maximum value of 100.
- To force clustering using a distance matrix based comparison, check Use distance matrix instead of k-tuple based mbed method for initial alignment.
The following table shows how the MegAlign Pro settings correspond with settings in Clustal Omega online.
| MegAlign Pro setting | Clustal Omega setting |
|---|---|
| Iterate to refine alignments | Does not correspond with the command-line, but enables the two settings directly below. |
| Maximum number of cycles of tree refinement | max-guidetree-iterations=(integer) iterations=(integer) |
| Maximum number of cycles of refinement using alignment-based HMMs | max-hmm-iterations=(integer) iterations=(integer). The value used for “iterations” is the maximum of the values specified for this option and the option in the preceding row. |
| Use distance matrix instead of k-tuple (enabled) | full |
| Use mbed for first round and distance matrix for later iterations (enabled) | full-iter |
| Maximum sequences per subcluster | cluster-size=(integer) |
After making your changes:
- Choose Align to use the entered options to perform a multiple sequence alignment.
- Use the Reset to Default button if you would like to reset all values to the MegAlign Pro defaults.
- Select Cancel to leave the dialog without saving any changes or performing the alignment.
Need more help with this?
Contact DNASTAR