
To perform the initial multiple sequence alignment:
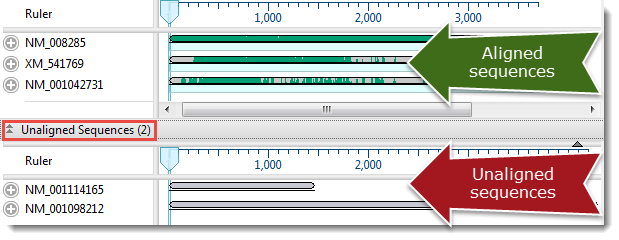
- Select three or more sequences to align, or select nothing if you want to align all sequences. (Note: If you choose just two sequences, or if only two are present, you will be prompted to instead do a pairwise alignment.) All sequences must belong to the same category (DNA/RNA or protein). Selected sequences can include any combination of already-aligned sequences and sequences in the “Unaligned Sequences” area. Both types are visible in the Overview (see image) and Sequences view.
- Hover the mouse over the Align/Realign tool (
![]() ) to view a tool tip showing the currently-selected algorithm.
) to view a tool tip showing the currently-selected algorithm.
- Choose the desired multiple sequence alignment method and perform the alignment. MAFFT [auto] is the default method for nucleotide alignments, MAFFT [l-ins-i] is the default for protein sequence alignments, and MAUVE is the default if the sequences are >1Mb in size.
- If the method and options are compatible with the current sequence type, and you want to use it, press the Align/Realign tool. In general, we recommend using Mauve for genome scale alignments and the other alignment methods for gene-level alignments. Since Mauve works by initially breaking alignment projects into a series of smaller sub-alignments, we also suggest using Mauve if there is insufficient memory to align sequences with the other methods, or if another type of alignment is taking an excessive amount of time to complete.
- To choose a different method, click on the small black arrow to the right of the Align tool to choose from available methods. Or use the main menu to choose Align > Align Using (Method Name). If the sequences have already been aligned, note that the menu command names change to Align > Realign Using (Method Name).
- To choose a different method and/or change method options before aligning sequences, click on the small black arrow to the right of the Align tool and choose Align with Options; or select Align > Align with Options from the main menu. For a detailed description of the Alignment Options dialog for different methods, see Clustal Omega, Clustal W, MAFFT, MUSCLE or Mauve alignment options.
- If the method and options are compatible with the current sequence type, and you want to use it, press the Align/Realign tool. In general, we recommend using Mauve for genome scale alignments and the other alignment methods for gene-level alignments. Since Mauve works by initially breaking alignment projects into a series of smaller sub-alignments, we also suggest using Mauve if there is insufficient memory to align sequences with the other methods, or if another type of alignment is taking an excessive amount of time to complete.
Except in the case of divergent sequences (see note, below), we recommend using the DNASTAR default settings the first time you perform a multiple sequence alignment. Later, you may wish modify some of the Alignment Options before realigning the sequences. After you customize options in the Alignment Options dialog and perform an alignment using the new settings, they become the new defaults, but only for this project, and only as long as the project remains open. For subsequent alignments in the same project, you can use the Align tool ( ) to perform the alignment using your custom settings.
) to perform the alignment using your custom settings.
- (optional) During the alignment, MegAlign Pro displays a progress window. If you need to interrupt the alignment, click Cancel. To view a console window showing the start time and progress of the alignment, click Show Console.
- If an alignment fails, you will receive a message with recommendations on how to obtain a successful alignment (e.g., by modifying options or choosing a different alignment method). See Troubleshooting ‘wrong type’ or ‘too long’ errors or Troubleshooting ‘fatal error’ in Clustal Omega alignments.
- If an alignment finishes successfully, a table of pairwise distances and a phylogenetic tree are created and displayed in the Distance view and Tree view, respectively. If the Console is not already open, and you wish to view alignment statistics and other information there, select View > Console. To reset the sequences to the unaligned state (i.e., as if they were just added to MegAlign Pro), use Align > Unalign All.
- If an alignment fails, you will receive a message with recommendations on how to obtain a successful alignment (e.g., by modifying options or choosing a different alignment method). See Troubleshooting ‘wrong type’ or ‘too long’ errors or Troubleshooting ‘fatal error’ in Clustal Omega alignments.
Need more help with this?
Contact DNASTAR