
In this tutorial, you will use an .abi read file to close a gap between two contigs.
- Download T6_Gap_Closure.zip (45 kb) and extract it to any convenient location (i.e., your desktop). The data set consists of one very small SeqMan Ultra project and one .abi file.
- Launch SeqMan Ultra and use File > Open to open the file De novo assembly.sqd. This assembly contains two unscaffolded contigs
- From the Explorer panel, use Shift+click to elect both contigs. Then right-click on the selection and choose New Scaffold.
- Select the scaffold (“Scaffold 1”) and press the Show Strategy view tool (
![]() ) to the right of the selection.
) to the right of the selection.
 ) to the right of the selection.
) to the right of the selection.In the Strategy view, note that each contig is comprised of two reads and that there is a clear gap between the two contigs.

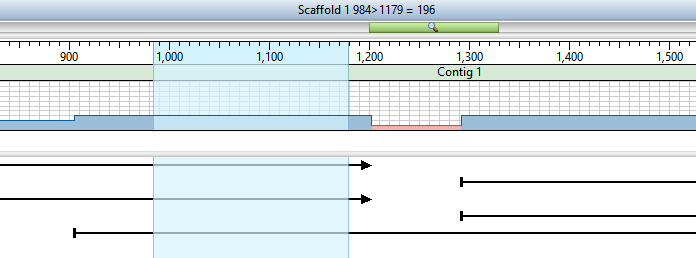
- Drag the cursor across the ruler area to select a range that spans the gap between the two contigs. There is no need to make a precise selection.

- Right-click on the selection and choose Add Sequences to Close Gap.
- Click the Add button and add the sequence Gap filling read.abi. Note that there are changeable Alignment options, but leave them at the defaults for purposes of this tutorial.

- Click Run. At the bottom of the report, observe the text: MERGING Contig 1 with Contig 2: percent match 99 (or 100).

- Click Finish
- Return to the Strategy view and observe that the gap is now closed. In the image below, note that the selection is the same as in Step 5.
Need more help with this?
Contact DNASTAR