
This part of the tutorial demonstrates that if you don’t have annotations to rely on for validating multiple alignments, a pairwise alignment is a good alternative.
This tutorial uses a default MUSCLE alignment of the Drosophila melanogaster ADH gene and the mRNA transcripts for isoforms F,C,E and H. In this example, some of the annotations of the original GenBank entry, NT_033779.5, were removed for the sake of clarity.
To begin the analysis:
- If you have not yet downloaded and extracted the tutorial data, click here to download it. Then decompress (unzip) the file archive using the method of your choice.
- Double-click on the DmADH.muscle.msa project file to launch it in MegAlign Pro.
The Overview for this alignment is shown in the image below. (Click on the image to make it larger.)
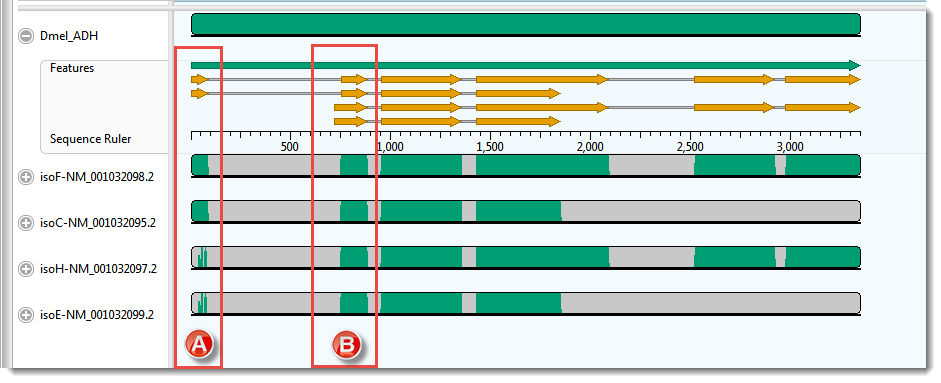
In this example, the transcripts, represented by the four lower sequences, have already been arranged in the same vertical order as the mRNA-features for the ADH gene, represented by orange arrows in the upper sequence.
- Hover your mouse over some of the orange mRNA features and verify that they are in the same order as the transcripts represented by the lower four sequences.
As expected, there is a good correspondence of the aligned regions of the transcripts to the exons of the gene. But a closer examination reveals a few problems, one of which is indicated by the red boxes in the image above. Notice that that isoF and isoC are annotated to code for Exon I (box A), which matches the multiple alignment result. However the 5’ end of isoH and isoE are also aligned, somewhat poorly, to this exon. How can you tell? Note that there are only two yellow arrows in box A, but there are green bars in all four transcript rows for box A, the bottom two of which are much sparser than the others This part of the lower two transcripts — which have no corresponding yellow arrows — should really have aligned with the start of Exon II (box B).
As a reminder, the example above used the MUSCLE alignment method. Before testing whether or not pairwise alignment can resolve this problem, check out how some of the other three multiple alignment engines handle these particular sequences.
- Multiply realign the sequences using Clustal Omega (Align > Realign Using Clustal Omega).
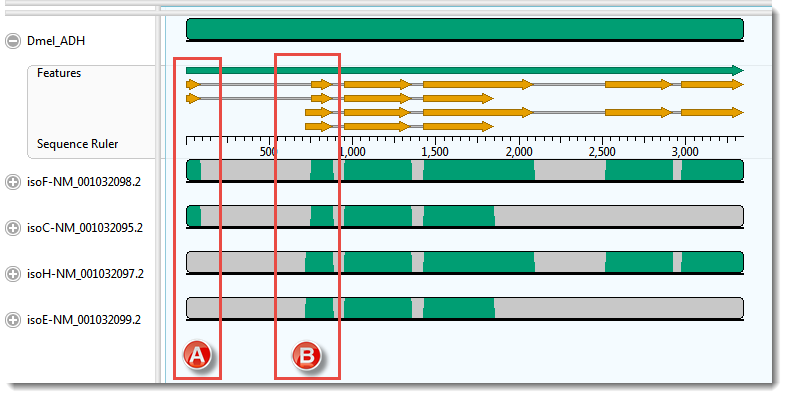
- Examine the alignments of transcript sequences to the first two exons of the gene’s sequence. In this case, there are no green bars at all in box A.
- Realign again, this time using MAFFT (Align > Realign Using MAFFT). For this example data set only MAFFT gives a result that is consistent with the annotations. (Click on the image to make it larger.)
Proceed to Part B: Use a Local pairwise alignment method.
Need more help with this?
Contact DNASTAR