
This tutorial demonstrates a situation in which a pairwise alignment can help resolve a confusing placement of gaps within a multiple alignment. In this case, a multiple protein sequence alignment suggests that the protein sequence from a specific organism (Tupaia chinensis, the Chinese tree shrew) is severely truncated at its C-terminus and ends with a run of 27 residues that seem to be unrelated to the other members of the alignment, including a relative (Sorex araneus, the Eurasian shrew). A pairwise global alignment of the two shrew sequences, however reveals that a more likely interpretation is that the T. chinensis sequence contains a deletion of 235 residues followed by a terminal stretch of 32 amino acids that is nearly identical to that of the S. araneus sequence. Here the pairwise alignment suggests that first-pass multiple sequence alignment is not optimal. Armed with this information, you can try changing the alignment engine and gap penalties to see if a more reasonable result can be achieved. Another technique that might help is to use sub-alignments to refine the overall multiple sequence alignment.
This example also demonstrates the power of using pairwise and multiple alignments together to help interpret specific relationships between sequences that might have become obscured by gaps which were added during the multiple alignment process. With MegAlign Pro, it is simple to generate many pairwise alignments without ever having to disturb the multiple sequence alignment, which has the larger picture. This is far more convenient that starting over with several different documents.
To perform the analysis:
- If you have not yet downloaded and extracted the tutorial data, click here to download it. Then decompress (unzip) the file archive using the method of your choice.
- Double-click on the Angiomotin_vertebrates.clustalo.msa project file to launch it in MegAlign Pro.
This project contains a collection of Angiomotin proteins from a diverse set of vertebrate species, already multiply-aligned with Clustal Omega. The figure below shows the alignment with the Sequences view and Overview scrolled to show the two tree shrew sequences. For more detail, you can expand the image below by clicking on it.
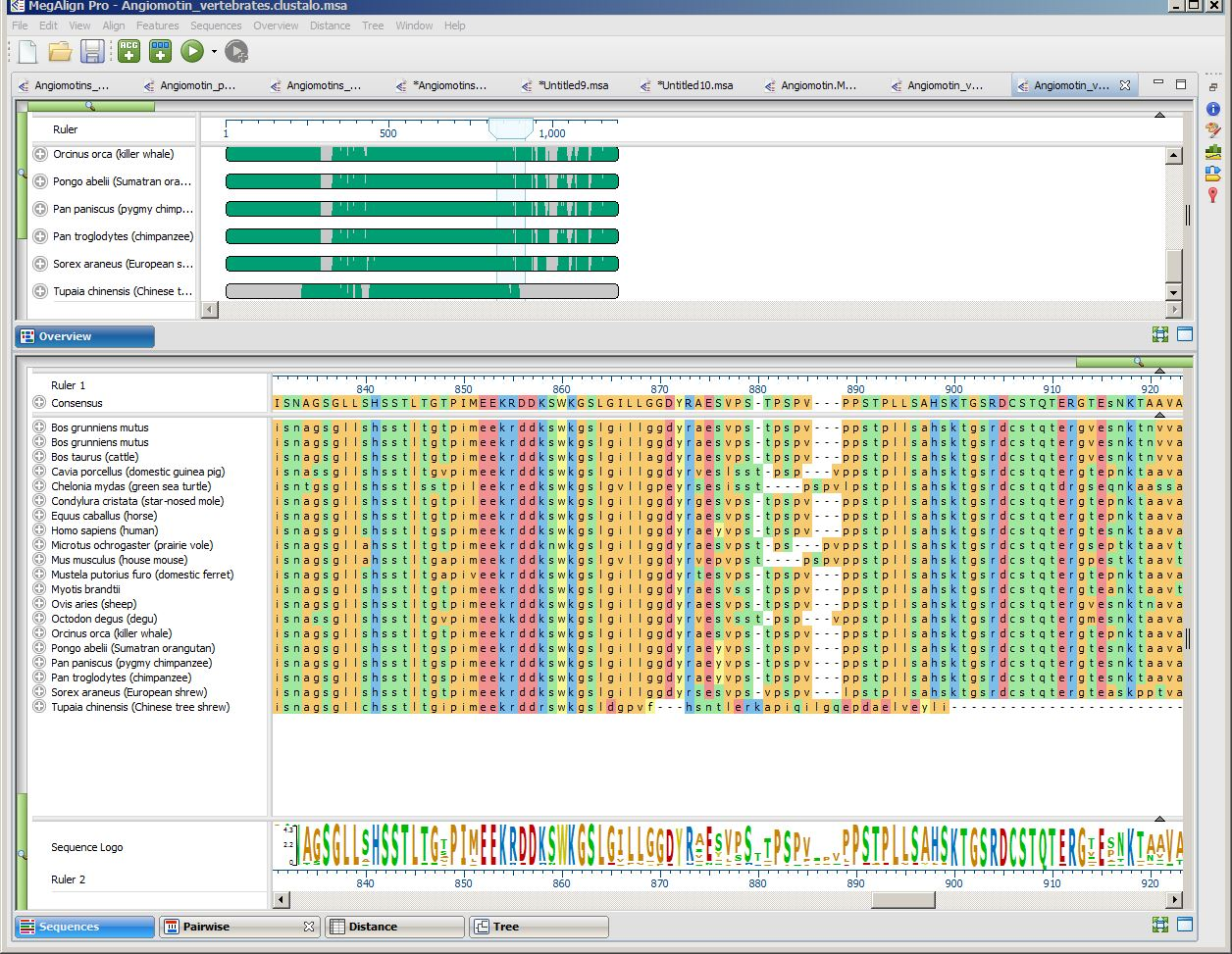
- Notice that the Tupaia sequence (the lowermost item on each list) seems to end abruptly with an unaligned run of amino acids. If you can’t see the abrupt end, move the Sequence view’s green horizontal zoom slider to the left until the Sequences view appears as in the image above.
- To investigate this using pairwise alignment, click on the name of the Tupaia sequence to select it, and then Ctrl-click (Win) or Cmd-click (Mac) on the name of the Sorex sequence to add it to the selection. You can select these sequences in either the Sequences view or the Overview.
- Right-click on either name and select Align Pairwise.
- In the Align Pairwise dialog box, select Global: Needleman-Wunsch from the Using drop-down menu. Since you are specifically interested in the C-terminal ends of the sequences, the Global option is ideal.
- Keep the default settings for all other dialog options and click OK. The alignment will run and a Pairwise view will appear.
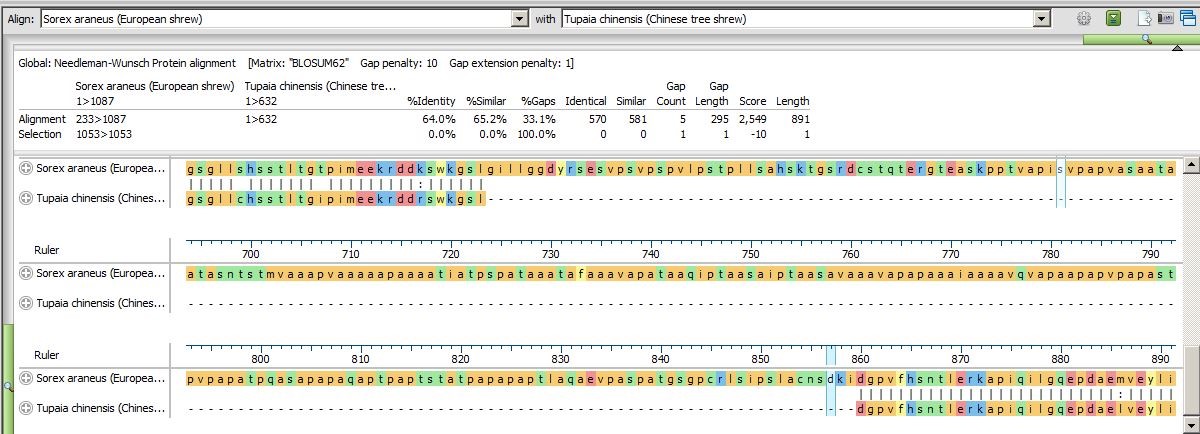
- Scroll down the Pairwise view to the end of the alignment and notice that it is now clear that the Tupaia sequence contains a large deletion and that the C-terminal ends of the two shrew sequences are nearly identical.
Pairwise alignments can also be used to help interpret multiple alignments with gaps that might obscure the relationships between sequences. While the Pairwise view is still open, try some Global alignments between different species pairs. This is easily done by changing the sequence(s) in the drop-down menus at the top of the Pairwise view.
- Change the sequence in the right drop-down menu to to Homo sapiens (human).
- Scroll through the updated alignment and observe that the two sequences match pretty well, despite some mismatches and gaps. The header shows a similarity calculation (Similar) of 91.3.
- Change the sequence shown in the left drop-down to Pan troglodytes (chimpanzee). The human and chimp sequences are 98.4% similar, with only an11-residue gap differentiating them. This similarity can be seen with a much greater degree of clarity than could be seen looking at the Overview of the original multiple alignment. (Click on the image below to expand it.)
Need more help with this?
Contact DNASTAR