
Individual sequences in the project can be trimmed before or after alignment based on features or by direct selection. This is different from trimming the entire alignment, which affects all sequences in the project.
To trim an individual sequence before or after performing an alignment:
Select a sequence in the Overview or Sequences view, then do any of the following:
- Press the Trim Sequence tool (
![]() ) from the toolbar.
) from the toolbar.
- Choose Sequences > Trim Sequence.
- Right-click on the sequence and choose Trim Sequence.
The Trim Sequences dialog opens and the sequence range text boxes are initially populated with the first and last nucleotides or amino acids.
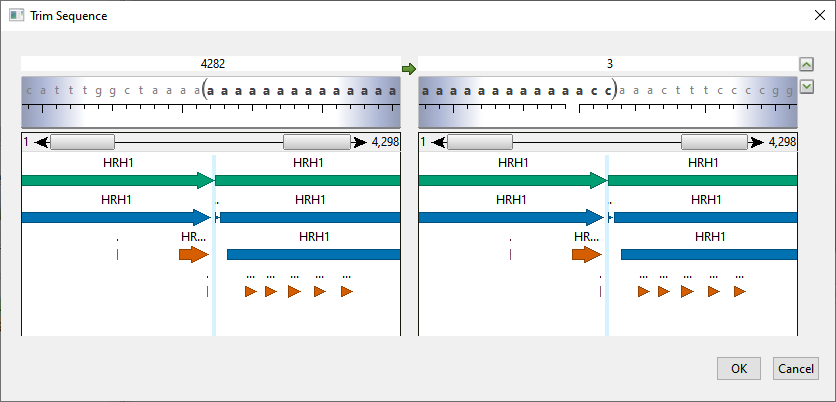
The following table shows tasks that can be done from within this dialog, or that affect its appearance.
| Task | How To |
|---|---|
| To reveal/hide the sequence, ruler, and any features that are present | Click the up/down arrows to the right of the range textboxes. |
| To specify the desired range | Use the “Left end” and “Right end” boxes to specify the desired range using any of these methods.
|
| To skip to the left or right ends of the sequence | Click in a range text box, then use the left/right arrows that appear within the box. Alternatively, type in “lend” (left end) or “rend” (right end) without quotes. |
| To view feature information | Hover above a feature (if any are present) to see information about it, such as its left and right coordinates. |
| To reverse complement the current sub-range | Click the green arrow between the two range textboxes. |
IUPAC ambiguity codes are recognized both in the sequence and in the range boxes. For example, typing AAS (where S = C or G) into a range box would cause SeqNinja to look for the first instance of AAC or AAG in the sequence. Conversely, typing AAC into a range box would cause SeqNinja to look for the first instance of AAC, as well as any combination of bases and ambiguity codes that would allow for AAC (e.g., AAS, WWM, etc.).
Need more help with this?
Contact DNASTAR