
When setting up a NovaFold prediction in Protean 3D’s NovaFold wizard, the second screen is Options. This screen allows you to edit or specify prediction options. During the prediction step, options specified in this screen are applied to all jobs listed in the Sequences screen.
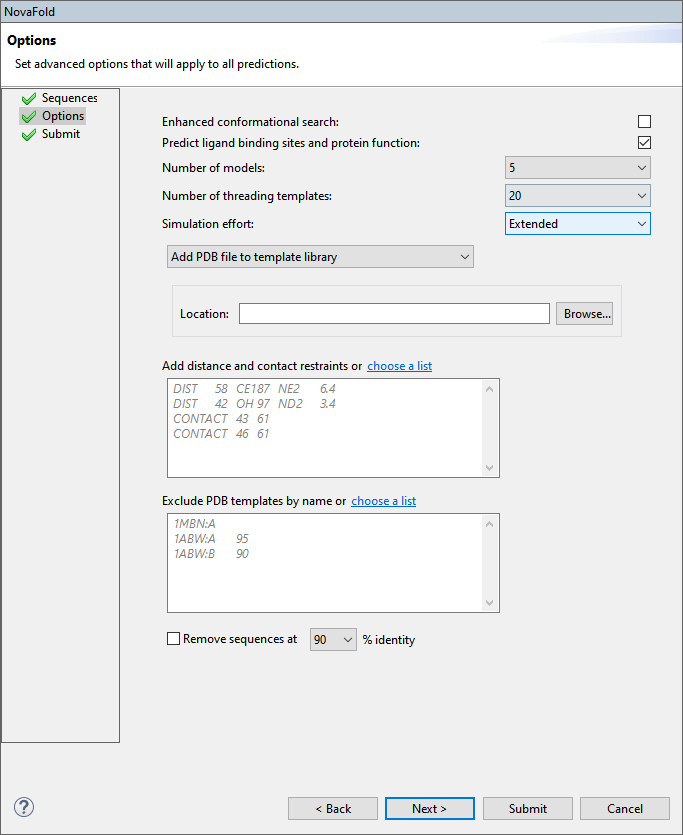
Edit or enter information in this wizard screen as desired. Or you can accept the defaults as they are, and simply press Next to continue to the Submit screen.
- Enhanced conformation search – If this box is checked, NovaFold will perform DNASTAR’s experimental method for enhancing the structural diversity of the normal template set. For templates selected by protein threading, a proprietary process samples alternate structural conformations and replaces a subset of the templates with lower energy conformations. This box is unchecked, by default.
- Predict ligand binding sites and protein function – If this box is checked, NovaFold will perform structure/function relationship calculations in addition to the folding prediction. These calculations are designed to elucidate a possible structural mechanism for a putative biological function. Uncheck the box if you do not need this additional prediction information and/or to obtain a faster folding prediction.
- Number of models – Select the maximum number of models to create [1, 5]. The default is 5.
- Number of threading templates – Select the maximum number of templates to use for simulations [1, 50]. The default is 20.
- Simulation effort – Specify whether to run Quick (5 hour) or Extended (50 hour) simulations. The default is Extended.
- The fourth drop-down menu on this screen lets you choose from the following options:
- Add PDB template by name – To nominate a PDB entry to use as a template in the modeling prediction, along with other templates selected by NovaFold. This option requires you to type in a four digit alphanumeric PDB code, followed by a colon and the chain name. For example: 4HHB:
- Add file with your sequence aligned to PDB template – Select this radio button to provide a text file containing a user-defined template structure and the alignment between that template and the query sequence. Then press Browse and select a text file containing either the pairwise FASTA-formatted sequence alignment between query and template, or the standard PDB format 3D structural coordinates of a single protein chain. See an example file further down this topic.
- Add PDB file to template library – Select this radio button to nominate a local 3D structure to use as a template in the modeling prediction, along with other templates selected by NovaFold. Then press Browse and select a text file representing a single protein chain in standard PDB format.
- Add PDB template by name – To nominate a PDB entry to use as a template in the modeling prediction, along with other templates selected by NovaFold. This option requires you to type in a four digit alphanumeric PDB code, followed by a colon and the chain name. For example: 4HHB:
This menu is followed by a Location box. Use the Browse button to navigate to the desired file location.
- Add distance or contact restraints – To restrict predictions based on distance and/or contact restraints (e.g., active sites, zinc fingers, disulfide bonds), type the information into the text box provided. This information will consist of a list of pairwise distances between two atoms (“i” and “j”), or contacts between two residues (“i” and “j”). Alternatively, click on the “choose a list” link to browse to a text file containing the same information. Both distances and contacts can be described in different rows of the same restraint file. IMPORTANT: The text file may not contain any lower-case letters. See an example file further down this topic.
- Exclude PDB templates by name – To exclude specified PDB structures from being used as templates for the prediction, type their name and chain information into the text box. Alternatively, click on the “choose a list” link to browse to a text file containing the same information.
- Remove sequences at ___% identity – Check this box to remove structural templates at or above the specified sequence identity from consideration. This setting was designed primarily for DNASTAR’s internal benchmarking. However, it is also useful for reducing the influence of closely homologous templates. For example, you could select 100% to exclude perfect matches.
Click Next to proceed to the Submit screen, or Back to return to the Sequences screen.
——————————————————————————————————————
Example text file for Add file with your sequence aligned to PDB template:
The following is a file for mammoth myoglobin (query) against whale myoglobin (target structure). “ATOM” rows 6-1211 have been omitted for space. The format for ATOM records is described on this PDB web page.
>query
MGLSDGEWELVLKTWGKVEADIPGHGLEVFVRLFTGHPETLEKFDKFKHLKTEGEMKASE
DLKKQGVTVLTALGGILKKKGHHQAEIQPLAQSHATKHKIPIKYLEFISDAIIHVLQSKH
PAEFGAD---------------------------
>1MBN:A
-VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASE
DLKKHGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRH
PGDFGADAQGAMNKALELFRKDIAAKYKELGYQG
ATOM 1 N VAL A 1 -2.900 17.600 15.500 1.00 0.00 N
ATOM 2 CA VAL A 1 -3.600 16.400 15.300 1.00 0.00 C
ATOM 3 C VAL A 1 -3.000 15.300 16.200 1.00 0.00 C
ATOM 4 O VAL A 1 -3.700 14.700 17.000 1.00 0.00 O
ATOM 5 CB VAL A 1 -3.500 16.000 13.800 1.00 0.00 C
…
ATOM 1212 NE2 GLN A 152 -1.600 24.200 -1.500 1.00 0.00 N
ATOM 1213 N GLY A 153 1.500 24.700 -6.400 1.00 0.00 N
ATOM 1214 CA GLY A 153 1.100 24.000 -7.600 1.00 0.00 C
ATOM 1215 C GLY A 153 0.300 22.700 -7.500 1.00 0.00 C
ATOM 1216 O GLY A 153 -0.900 22.800 -7.100 1.00 0.00 O
TER 1217 GLY A 153
Example text file for Add distance and contact restraints:
DIST 12 HG21 50 HB1 8.1
DIST 14 HA 57 1HE 6.2
DIST 21 HB2 43 HD11 4.0
DIST 124 CA 84 CA 17.4
DIST 36 UNK 120 CA 17.4
CONTACT 33 6
CONTACT 60 29
CONTACT 37 345
CONTACT 109 42
Column requirements in the text file:
Given two residues that contact one another (‘Residue i’ and ‘Residue j’) or two atoms at a distance from one another (‘Atom i’ and ‘Atom j’:
| Column name | Description |
|---|---|
| Distance rows contain the following columns from left to right: | |
| “DIST” (without quotes) | |
| Res_No.i | Residue sequence number for Residue i |
| Atom_type_i | Atom name for contacting atom of Residue i |
| Res_No_j | Residue sequence number for Residue j |
| Atom_type_j | Atom name for contacting atom of Residue j |
| Distance in Angstroms | |
| Contact rows contain the following columns from left to right: | |
| “CONTACT” (without quotes) | |
| Res_No.i | Residue sequence number for Residue i |
| Res_No.j | Residue sequence number for Residue j |
In both cases, UNK can be used in a row to represent an unknown atom.
Need more help with this?
Contact DNASTAR