
STREAMLINED IDENTIFICATION AND CHARACTERIZATION OF VIRAL STRAINS

Chapter 1: General Procedure for Identifying and Characterizing Viral Strains
What is the general procedure for identifying and characterizing viral strains?
There are four main steps involved in identifying and characterizing viral strains.
The first step happens even before you send your samples to be sequenced.

To download this entire ebook as a PDF, click here.
Step 1: Decide whether you will be assembling your experimental sequence reads to a reference or assembling de novo
Before deciding on a sequencing technology (Step 2), it is important to know whether you will be assembling your sequences de novo or against a reference sequence.
If you are working with a novel virus, or if your virus has a rapid mutation rate or large-scale changes (as seen with segmented reassortment), a closely matching reference genome will not be available. For these viruses, there is no choice but to do de novo assembly.
For more stable viral genomes, where the mutation rate is relatively slow, a genome reference sequence from the same (or closely-related) strain can be used to guide alignment of the sequencing reads. If a reference sequence is available for your virus of interest, we highly recommend that you use it. Templated assembly helps order contigs into larger scaffolds, giving you a more complete genomic sequence, and allows for both faster and more accurate assembly. These assemblies are ideal for monitoring the evolution of viral strains such as SARS-CoV-2. Downstream analysis typically focuses on identifying variations that define a viral strain or affect viral proteins.
Step 2: Choose the sequencing technology and submit your samples
If you will be doing a reference-guided assembly, Sanger or Illumina sequencing technologies are perfectly suitable for viral genome analysis. These technologies create shorter reads than some of the more recent technologies but are still highly accurate.
If you will be doing de novo assembly, the sequencing technology you choose will depend on the length of the viral genome you are studying. Excellent de novo assemblies for viral genomes up to 10Kbp in length can be produced with Sanger and Illumina 2X300bp MiSeq reads. However, accurate de novo assembly of long and/or more complex genomes like SARS-COV-2 (30Kbp) or influenza (segmented genome) is best accomplished using long reads.
Long-read sequence data provides many advantages to earlier sequencing types. At the time of this ebook’s publication, there are three main long-read sequencing technologies: Oxford Nanopore Technologies (ONT/Nanopore), Pacific Biosciences (PacBio) CLR, and PacBio HiFi.
PacBio HiFi sequencing, in particular, produces long and accurate (>99.9%) sequence reads that work well for both reference-guided and de novo assembly. PacBio HiFi reads are produced via PacBio’s Sequel HiFi sequencing platform using a hybrid short and long read sequencer mode called “circular consensus sequencing” (CSS). PacBio HiFi reads are shorter but more accurate than long-reads produced using the other two long read sequencing technologies. Due to this high level of accuracy, viral genome data sequenced using PacBio HiFi technology often assembles into a single contig.
Once you’ve decided on a sequencing technology, sequence your experimental samples in-house or submit them to a core facility. If you have chosen a long read technology, note that most small sequencing facilities do not have access to long-read sequencing platforms. To obtain reads in these formats, you may need to send your samples to a large outside facility.
Depending on the facility, you will get back either raw sequence reads or draft genomes.
If you will be starting with raw sequence reads, proceed to Step 3, where you will assemble those reads into draft genomes. If you already have draft genomes, instead go to Step 4, where you will analyze evolutionary relationships and find sequences and variants of interest.
Step 3: Assemble raw reads into a set of draft genomes
If you are starting with raw sequencing reads, you will need to use software to assemble them into a set of draft genomes. A couple of decades ago, this step could take up to an hour. Today, if you are using long reads that span the entire viral genome, modern software can complete your assembly in as little as a few seconds.
For de novo assemblies, you won’t need anything other than the assembly software and sequence read file(s), typically in FASTQ format. The assembly software will produce one or more contigs that represent your viral draft genome. We recommend choosing software that allows you to visualize and edit these contigs in order to produce the most accurate draft genome. This draft genome can then be exported to FASTA format and compared to other viral strains in Step 4.

If you have a closely related reference genome, you can use that to guide assembly of your raw sequencing data. The National Center for Biotechnology Information (NCBI) is an excellent source of reference genomes that can be downloaded for free. Click here to visit NCBI’s nucleotide reference genome search page. Variants between the sequenced strain and reference genome can then be detected and written to a variant report or VCF file for further analysis in Step 4.
Step 4: Compare the experimental draft genomes to one another and/or to reference sequences
If your experimental draft genomes were assembled using a reference, you will likely be interested in finding important variants. Look for software that supports filtering so you can limit found variants to only the SNPs and indels of interest. The ideal software should accommodate common file formats for variant calls, like VCF, and should also allow you to easily compare variants across multiple samples using specific filtering criteria.
Multiple sequence alignment can be used to compare draft genomes to one another or to related reference sequences. When aligning large numbers of viral genomes, there are many algorithms available. However, MAFFT7 is considered to be superior for large numbers of viral genomes due to its accuracy and high capacity.
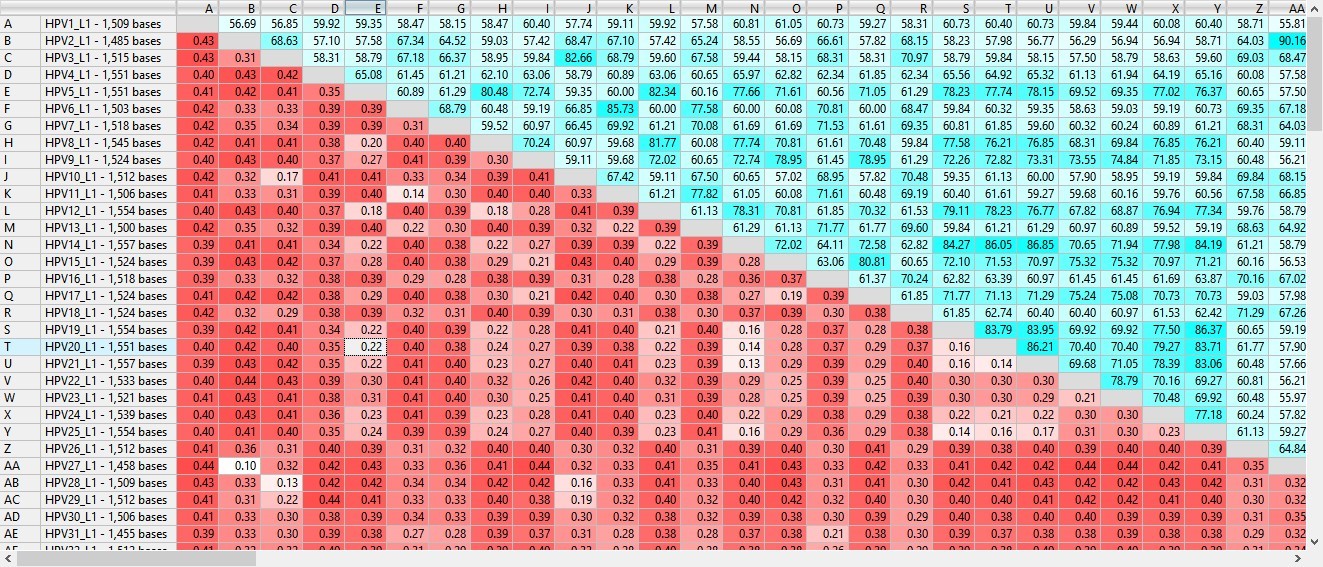
Figure 1. Section of a MegAlign Pro distance table for 10,000 viral samples aligned with MAFFT.
After multiple sequence alignment, the results can be viewed in the form of a distance table (Figure 1) or a phylogenetic tree. If you want to home in on the differences between two strains, some software lets you align just those two sequences using an algorithm optimized for pairwise comparison.
See the box on the following page for one example of how to use multiple sequence alignment to identify a viral strain of SARS-CoV-2 in an experimental subject.
Example: Identifying the strain of an experimental virus sample using a phylogenetic tree
This example is taken from a DNASTAR tutorial using MegAlign Pro multiple sequence assembly software.
First, an experimental viral sequence (SRR13380669_ NC_045512.2) from a subject with an unknown strain of SARS-CoV-2 was uploaded to a multiple sequence alignment application. Reference sequences from NCBI for four known strains of SARS-CoV-2 were also uploaded (see table).
Next, the MAFFT algorithm was chosen, and a multiple sequence alignment was performed using all the uploaded sequences.
A phylogenetic tree was automatically generated from the resulting alignment (Figure 2). In the image below, the experimental sample SRR13380669_NC_045512.2 is shown as part of the clade with a green background, B-1-351, or the “Beta” strain or SARS-CoV-2.
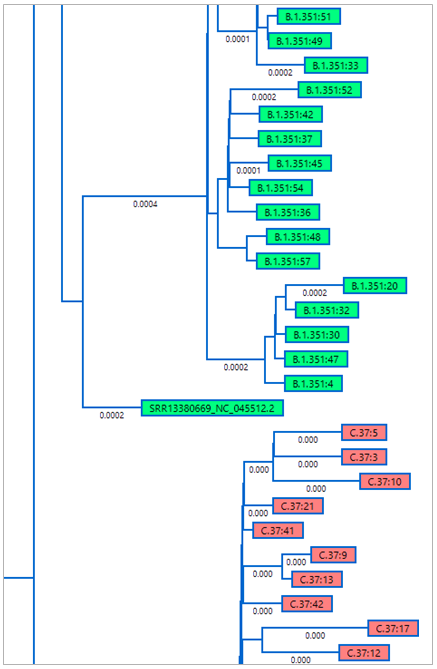
Figure 2. Portion of the MegAlign Pro phylogenetic tree showing placement of the experimental sample in the “Beta” strain clade.
To download this entire ebook as a PDF, click here.
As verification, the distance table for the alignment was automatically ordered from most- to least-related to the experimental sample (Figure 3). The most-related samples were all from the “Beta” strain, offering further confirmation that SRR13380669_NC-045512.2 was indeed from that strain.
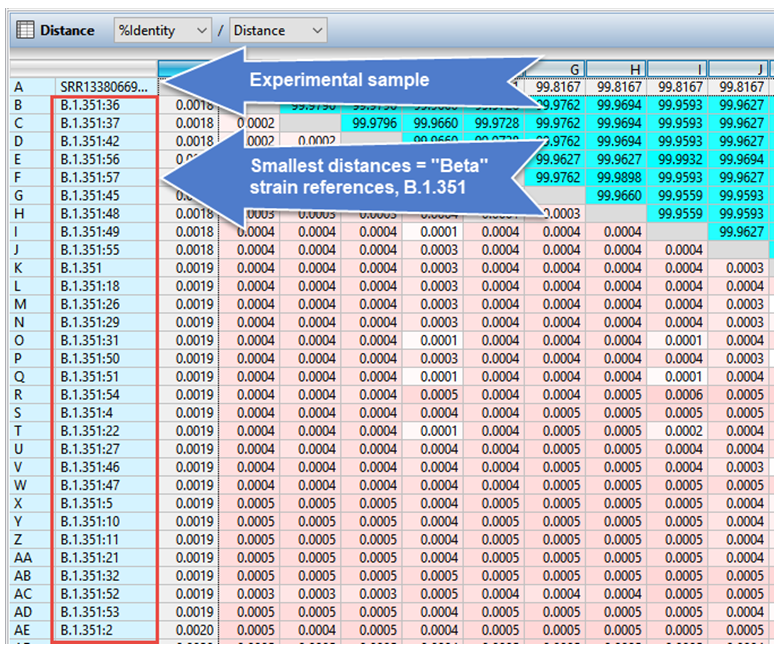
Figure 3. Portion of the MegAlign Pro distance table confirming placement of the experimental sample in the “Beta” strain clade.