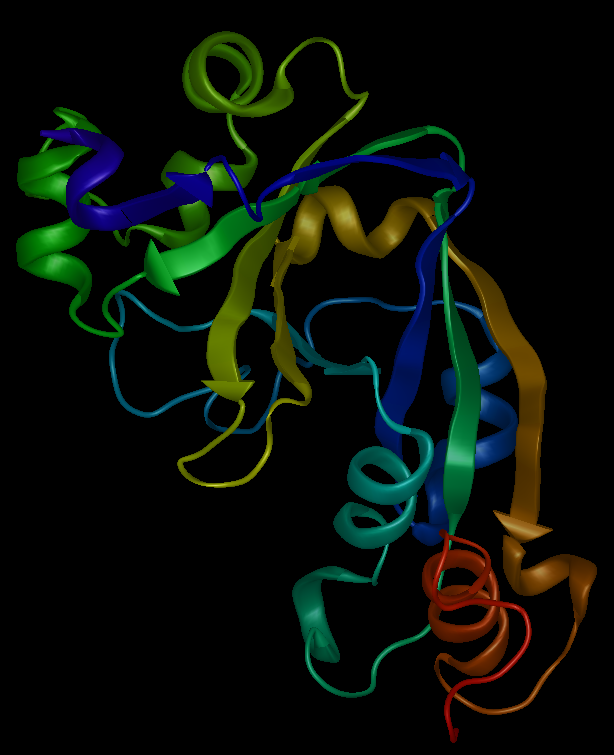
 Protein structure prediction software programs seek to solve one of the more essential bioinformatics quandaries: how can we determine the three-dimensional structure of a protein from its amino acid sequence? Important for both medicine and biotechnology, the task of protein structure prediction remains a complex question. All protein structure prediction algorithms utilize one or more of the following three approaches:
Protein structure prediction software programs seek to solve one of the more essential bioinformatics quandaries: how can we determine the three-dimensional structure of a protein from its amino acid sequence? Important for both medicine and biotechnology, the task of protein structure prediction remains a complex question. All protein structure prediction algorithms utilize one or more of the following three approaches:
Homology Modeling
The underlying assumption in the homology modeling approach is that proteins with similar amino acid sequences will share similar structures. The homology modeling approach maps the amino acid sequence from the protein you would like to predict (the target) onto the experimental structure of a closely homologous protein (the template). The candidate templates are identified by sequence alignment before the query sequence is then mapped onto the template scaffold. The homology method relies heavily on high sequence identity between the target and the template for accurate models. Inaccuracies in homology models can arise from errors in sequence alignments or splice variants in protein sequences.
Threading (Fold Recognition)
The protein threading method, also known as fold recognition, differs from homology modeling in that it can identify sequences with similar protein folds that exhibit less sequence similarity. In the case of threading, candidate templates are identified by profile alignment methods that consider both sequence and structural similarity. Some of the factors determining structural similarity include predicted secondary structure and predicted solvent accessibility. After the candidate templates are identified, the query sequence is mapped onto the template scaffold. The threading method will find both homologous structures as well as structurally similar ones to create the final prediction models.
Ab initio (Template Free) Protein Modeling
This type of modeling works to build proteins from scratch, rather than using solved structures as in the homology or threading methods. It relies on biophysical protein principles to create protein models, and requires immense computational resources. Originally known as ab initio prediction, the terminology for this type of modeling has been drifting recently. Rather than referring to physics-based methods (e.g. modeling the folding process) this method is now often called “template-free.”
DNASTAR Protein Structure Prediction Algorithms Use a Hybrid Approach
NovaFold uses a combination of both the threading and ab initio methods to perform protein structure predictions. In NovaFold, the threading method finds structure fragments from multiple templates and the ab initio method builds structure for regions not matching the template. By combining these two methods with the power of Amazon Web Services, scientists are able to obtain highly accurate structures using a robust protein structure prediction workflow in the most efficient and cost-effective manner possible.
NovaFold AI uses an artificial-intelligence based hybrid method that is radically different from I-TASSER. NovaFold AI incorporates the award-winning AlphaFold 2 AI system developed by DeepMind. AlphaFold 2, which uses threading and other methods, was the top-ranked protein structure prediction method in the CASP14 challenge in 2020, significantly outperforming the other participating teams by determining the structure of proteins with accuracy comparable to laboratory experiments.


2 Comments
Leave your reply.