
LASERGENE 17.2 RELEASE NOTES




Date: 9/30/2021 Version Affected: Lasergene 17.3.0.58 and earlier Version Fixed: Lasergene 17.3.0.59 and later Issue...
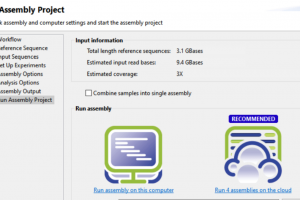
Check out our new post featuring questions and answers from our recent webinar on genomic...
MADISON, Wisconsin – July 18, 2013 – DNASTAR® announces the[...]
© 2024 — DNASTAR Privacy Policy
Leave a Reply
Your email is safe with us.