Who uses RNA-Seq transcriptome data? Molecular biologists, clinical researchers, bioinformaticians, geneticists, statisticians, computer scientists and anyone interested in differential gene expression and/or transcriptome variation.
Regardless of your scientific discipline, transcriptome projects can be complex. In this post, DNASTAR’s Manager of Sales and Client Support, Carl-Erik Tornqvist PhD, will answer some common questions about RNA-Seq analysis, and especially those pesky normalization methods.
Because we’d like this post to be a resource to students and researchers of all backgrounds, Carl-Erik will use the first half of this post to answer general questions about how RNA-seq data differs from genomic data. In the second half, he’ll provide an in-depth look at RNA-seq normalization methods.
Jump to Part I: Where does RNA-Seq data come from, and how is it assembled?
Jump to Part 2: What do I need to know about RNA-Seq normalization?

Part I: Where does RNA-Seq data come from, and how is it assembled?
How does RNA-seq “transcriptome” data differ from DNA genomic sequence data?
A genome is the DNA sequence that includes gene encoding and non-encoding sequences. The sequence is a blueprint for all genes that may be expressed. With genomic data, you can see the gene sequences and any variants between samples. However, there’s no information about the expression of genes.
By contrast, a transcriptome contains the gene encoding sequences only. A transcriptome is a snapshot of the expressed genes, through sequencing of cDNA (from mRNA), under the conditions in which the biological sample was isolated. Transcriptome data does not contain all RNA. Instead, only those sequences that form transcripts—the mRNA sequences—are isolated for further study.

In the lab, how does collection of RNA compare to that of DNA?
Due to the fragility of RNA molecules, RNA extraction requires enhanced sterile and decontamination procedures compared to DNA extraction. With RNA extraction, the genomic DNA is purposely degraded using the enzyme DNase, whereas any trace of RNase must be removed and prevented.
Who does the sequencing? What format are the resulting files saved in?
Usually, a sequencing core facility or company will perform the sequencing, which may also include preparation of the sample libraries. Depending on the instrumentation, sequence read length, and number of samples, a sequencing run can last from less than an hour to several hours. The cost per sample can be reduced by using “multiplexing” to mix multiple samples in a single sequencing reaction.
Typically, the sequencing facility also performs post-processing of the sequencing data. This includes things like adapter removal and demultiplexing.
The post-processed sequences are saved as .fastq files, with paired-end data having two .fastq files per samples, denoted by the letters “F” and “R” in their filenames. A nice open-source software utility that allows you to see the quality of the sequences in a .fastq file is called FASTQC. With FASTQC you can see useful summaries of the data such as number of reads and average read-depth.
Why is de novo RNA-Seq assembly called “transcriptome” assembly? How does it differ from reference-based assembly?
Reference-based RNA-seq assembly aligns sequencing reads to a reference or template sequence. In DNASTAR’s SeqMan NGen application (image below), there are both NGS-based and long-read versions of this workfow, both called “RNA-Seq.”
By comparison, the goal of de novo transcriptome assembly is to find novel transcripts and their expressed genes without using a reference sequence. In SeqMan NGen, the “De novo transcriptome” workflow is listed under “De Novo Assembly.” During de novo transcriptome assembly, similar contigs are grouped together as if they are from the same gene. The name of the workflow refers to the resulting “transcriptome” that is generated from the assembly.
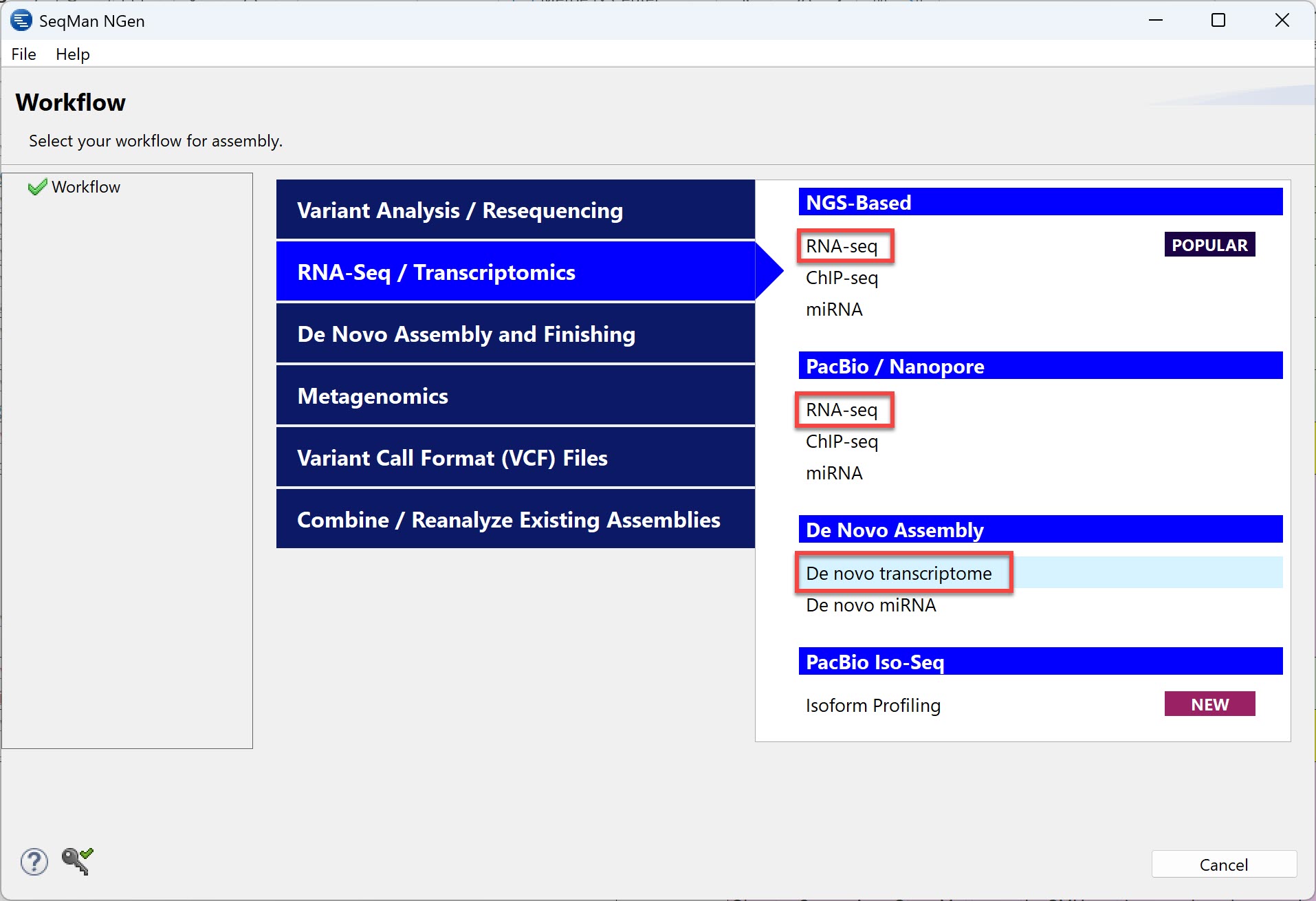
Using the following 2-step procedure, you can even use the same data set for both reference-based RNA-seq and de novo transcriptome:
1) Use the reference-guided RNA-seq workflow, then check the results (e.g., using DNASTAR’s SeqMan Ultra) to see which reads were unassembled. These reads are considered “novel” transcripts because they were not present in the reference sequence.
2) Assemble the “unassembled” reads de novo to see what additional transcripts are present in samples.
If I decide to run a reference-guided assembly, where do I get the template?
A template can be downloaded for free in any of the following ways:
- Use a licensed copy of DNASTAR SeqMan NGen and click a button on the “Reference Sequence” screen (see image).
– Use the Download Genome Package button to choose a genome by organism name from DNASTAR’s curated and up-to-date genome template database. This is the preferred reference source if you are working with a model organism, especially human, as these genome template packages are already annotated with Genomenon/Mastermind, dbSNP, and dbNSFP information. Then, when you explore the assembly results, you can view variants tables with links to the relevant web pages for each variant in addition to tables and views for gene expression analysis.
– Use the Download NCBI Genomes button to download and use a genome reference from the NCBI Entrez Genome Project database. Then use the drop-down menus provided to search by organism name or accession number.

If I follow the de novo transcriptome workflow, how can I recognize which transcripts are known and which are novel?
By comparing the found transcripts to those on NCBI’s RefSeq website. As a shortcut, DNASTAR’s SeqMan NGen provides a Transcript Annotation Database checkbox in its assembly wizard.
After checking the box, licensed users can choose from DNASTAR’s database of transcript annotations extracted from NCBI’s RefSeq database.

When setting up an RNA-Seq assembly, what can I do to ensure a clear signal in my RNA-Seq data?
Sample preparation is key. When you extract RNA from a sample, you collect the RNA from all sources present in that sample. If there is a known source of unwanted RNA in your sample, you can use contaminant scanning in the assembly to filter out the unwanted RNA sequences.
For example, if your sample is from a plant leaf, could that plant possibly have a virus? If your plant sample contains viral RNA and the sequence of that virus is available, some assembly software may allow you to automatically scan for the viral sequences and remove those reads prior to assembly, as DNASTAR’s SeqMan NGen does (see image). Also, ensuring that the total amount of isolated mRNA from each sample is equivalent will allow you to use more normalization approaches.

Assembly software like SeqMan NGen also allows you to remove universal adapter or scan for specific vectors/adapters prior to assembly.

Part 2: What do I need to know about RNA-Seq normalization?
What is data normalization and why is it used in RNA-seq assemblies? Are there other types of assemblies that also let you specify data normalization?
Normalization is a type of data standardization used to account for variations in the data. Normalization in RNA-Seq analysis is necessary to compare expression levels among gene transcripts of different lengths and to account for sample variation. Some normalization methods, such as DESeq2 and EdgeR, use statistical tests to assess differential expression. Though outside the scope of this post, normalization is also used for ChIP-seq, miRNA, and CNV analyses.
Normalization methods vary according to the software being used for assembly. The rest of this blog will discuss the RNA-Seq normalization methods offered in DNASTAR Lasergene software.
Which Lasergene applications support RNA-Seq data normalization?
Normalization methods can be selected through either SeqMan NGen or ArrayStar, both part of Lasergene Genomics. If you are starting a project in ArrayStar and are importing an experiment not assembled in SeqMan NGen, you would choose the normalization method in ArrayStar. However, if your starting point is to assemble your reads in SeqMan NGen, you would choose the normalization method in that application. This information is passed on to the ArrayStar file that is created automatically during assembly.

Which normalization methods are offered in ArrayStar and SeqMan NGen?
Regardless of whether you are using SeqMan NGen or ArrayStar, you will use a drop-down menu to choose from available RNA-Seq normalization methods.

- None: no normalization of the data
- Quantile: Normalization by distribution, in which all of the values in the project are adjusted so that the distribution is the same across all of the experiments. That is, each quantile is replaced by the average (or median) quantile across samples.
- RPM (Reads assigned Per Million mapped reads): Normalization by library size in which signal values for each experiment will be divided by the total number of mapped reads divided by one million.
- RPKM (Reads assigned Per Kilobase of target per Million mapped reads): Normalization by library size, in which signal values for each experiment will be divided by the total bases of target sequence divided by one thousand; and the resulting number divided by the total number of mapped reads divided by one million.
- DESeq2: DESeq2 analysis involves a statistical package in Bioconductor that uses a median of ratios method to normalize read counts. To test for differential expression, raw counts are used to fit a Generalized Linear Model of the negative binomial distribution.
- EdgeR: EdgeR differential expression involves a statistical package in Bioconductor that uses trimmed mean of M-values to normalize the read counts. To test for differential expression, normalized count data are used to estimate per-gene fold changes and to perform statistical tests.
Note that EdgeR and DESeq2 normalization methods require at least two replicates per sample and a control sample. If your input data does not include replicates, these normalization methods will not be available in the drop-down menu.
For an in-depth discussion of normalization methods available, see the user guides for SeqMan NGen and ArrayStar.
From the normalization methods available, how do I choose the best method for my data?
It depends on what you know about your data. If you know that the total mRNA per cell is equal, then normalization based on library size (e.g. RPKM) is an acceptable approach and can tolerate asymmetry in the differential expression, that is different numbers of genes can be up- or down-regulated across different conditions/samples. However, determining the amount of mRNA per cell is not an easy task, and this calculation is usually not performed.
Also, note that RPKM is not a good method for comparing between samples. It is best to use RPKM for within-sample comparison of different genes.
For experiments in which the total mRNA per cell is not equal among samples, then normalization by read count (DESeq2; edgeR) is an acceptable approach. However, normalization by read count performs poorly when there is a high degree of asymmetry in the differential expression across conditions/samples. Again, for the methods above, you will need at least two replicates per sample and one sample will need to be designated the control.
When might I NOT want to normalize my RNA-Seq data?
It is always good practice to normalize your data, however, if your data does not meet the assumptions that need to be met for a normalization approach, then you would not want to normalize the data and would choose None from the drop-down menu.
How can I tell if I chose the “wrong” normalization method? Are there any tell-tale signs when I’m doing downstream analysis?
If there is prior knowledge about the expression levels of certain genes called “housekeeping genes,” then you can analyze the expression levels of these genes across all samples, to evaluate the results. Housekeeping genes are genes that are considered necessary for cellular function and, therefore, would not show DE across all conditions. Housekeeping genes can also be used as controls in other normalization approaches.
Want to see these workflows in action? Watch a recording of our September 2020 webinar with Dr. Carl-Erik Tornqvist.
Ready to try Lasergene’s RNA-Seq workflows for yourself? Download a 14-day free trial or visit our workflow page to learn more.


1 Comment
Leave your reply.