
After finishing Part C, the protein structure with the variant of interest opens in Protean 3D.
In the Molecules section, two near-identical copies of the structure appear. The upper structure is the original structure from the Protein Data Bank (PDB). The lower structure is the variant version calculated by Protean 3D.
- Use the Structure view to observe the mutated side chain along the backbone. To show/hide the different versions of each chain, check/uncheck boxes in the Molecules section.
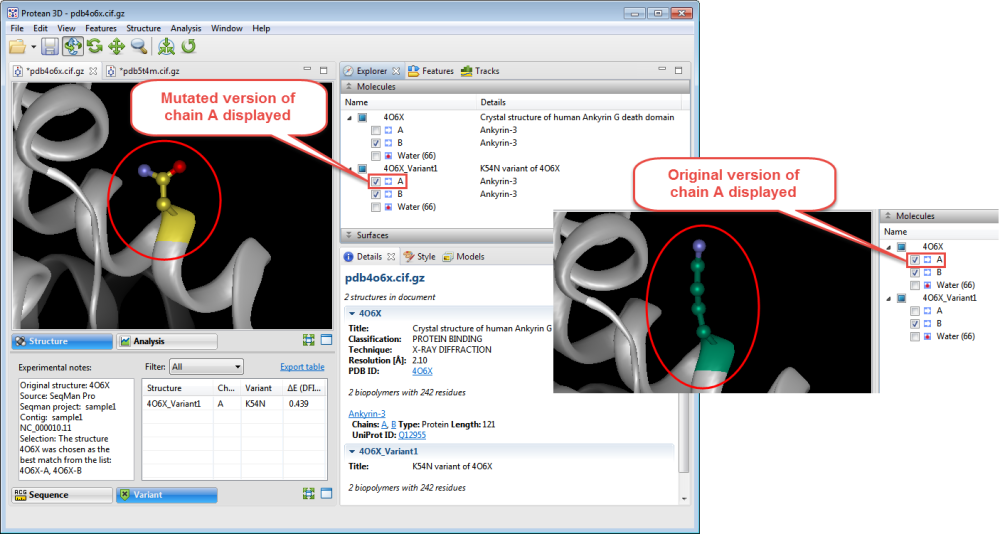
- To see notes about the structure chosen as the best match, look in the Notes box in the Variants report.

Protean 3D uses several metrics to determine the “best” PDB file to display when a variant is located in a CDS that is associated with more than one PDB file. Quality is the first consideration, with high-resolution crystal structures > NMR structures > low-resolution crystal structures > other techniques. This ordering is refined by alignment to the corresponding Uniprot sequences. This refinement considers the percent match and the number of gaps before the variant position. If two structures are still tied as the “best,” the largest structure is chosen.
- To predict whether the mutation is stabilizing or destabilizing to the protein structure, use the table on the right of the Variants view. The delta-E (DFIRE-A) column displays the change in energy value based on the DFIRE calculation (reference). This number can be used to predict whether the mutation is stabilizing or destabilizing to the protein structure. A positive number is considered destabilizing to the structure when compared to the original amino acid; a negative number is considered non-destabilizing.
- To further explore potential impact of mutation on protein stability and function, apply a solvent-accessible surface.
- In the Analysis view, analyze secondary structure characteristics to interrogate the mutation’s effect on protein flexibility, amphiphilicity, charge density, hydropathy, and more.
Need more help with this?
Contact DNASTAR