
The Protein Design reports, “Variants” and “Hot Spot Scan,” are two similar versions of the Report view. Both versions contain data that can be used to predict whether a SNP might be stabilizing or destabilizing to a protein structure. These reports are available in the following circumstances:
- A Protein Design result is open. By default, hot spot scan results cause the Hot Spot Scan report to open; variants results cause the Variants report to open.
- A variant from the the “SNP to Structure” workflow is open. In this case, instructions from SeqMan Pro cause Protean 3D to open two structures. One is the original structure from the Protein Data Bank, while the other is the variant version calculated by Protean 3D.
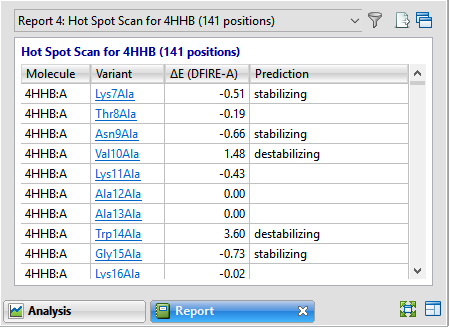
An example Hot Spot Scan report is shown below:
To open a Protein Design report, click on the Done link for a Protein Design job in the Jobs panel, use the View > Report > Show command or press the Report tab ( ). If the desired report is not currently displayed in the Report view, select it from the drop-down menu in the upper left corner of the Report view. If a Variants or Hot Spot Scan report is not in the menu list, add it using the Add reports to this view tool (
). If the desired report is not currently displayed in the Report view, select it from the drop-down menu in the upper left corner of the Report view. If a Variants or Hot Spot Scan report is not in the menu list, add it using the Add reports to this view tool ( ).
).
To open a variant structure from the report table, double-click its row.
To see experimental notes below the table, select a table row (molecule). To hide the notes, click the “Hide Notes* link above the table.
To learn about the tools available in this view, see Report view.
Interpreting Protein Design results:
The report table lists information about the variant structure(s). If desired, you can filter hot spot scan results.
| Table column | Description |
|---|---|
| Molecule | The structure and chain where the variant is located. |
| Variant | Each variant is shown in the form: [original amino acid] [position] [substitute amino acid]. Amino acids are shown using 3-letter IUPAC codes. In the Hot Spot Scan version of the report, each item in this column is hyperlinked. Click the link to open the Variants version of the report for the specified variant. |
| ΔE (DFIRE-A) | Displays the change in energy value based on the DFIRE calculation (reference). This number can be used to predict whether the mutation is stabilizing or destabilizing to the protein structure. A positive number > 0.5 is considered destabilizing to the structure when compared to the original amino acid; a negative number < -0.5 is considered stabilizing. Numbers between -0.5 and +0.5 are considered neutral. |
| Prediction | Stabilizing or destabilizing. Blank cells indicate a neutral prediction, neither stabilizing nor destabilizing. |
*Note: If the view pertains to a SNP-to-Structure result and the residue has no atoms in the PDB structure, you will see an error message similar to the one below. In this case, return to SeqMan Pro and choose a different row in the Variants table.

Need more help with this?
Contact DNASTAR